http://www.chemistrymag.org/cji/2003/057060pe.htm |
Jul. 1, 2003 Vol.5 No.7 P.60 Copyright |
Structure and electronic properties of C36
-rings Jin Yajuan, Huang Yuanhe*, Li Yuxue, Liu Ruozhuang(Department of Chemistry, Beijing Normal University, Beijing 100875, China)
Received Apr. 21, 2003; Supported by the National Natural Science Foundation of China (No.20273009) and the Major State Basic Research Development Program (Grant No. G2000078100).
Abstract Several new C36-rings
with Dnh symmetries have been constructed and calculated using
semi-empirical molecular orbital method (AM1). The ring structures are favorable to the
stability of the C36 systems. An analysis on several factors, such as the
change of strain energies due to the distortion of C36 cage, the position of
the bonded carbon atoms, the size of retained aromatic domains and the shared
pentagon-pentagon double bonds, is given for their contributions to stability of the C36-ring.
The electronic structures are also discussed.
Keywords C36-ring, semi-empirical molecular orbital method,
structure and electronic properties
1. INTRODUCTION
C36 cage has attracted much attention in recent years since it was
synthesized by arc-discharge method and purified in bulk quantities[1-3]. It is
known that C36 cage is highly reactive and has a strong tendency to form
intermolecular covalent bonds due to its diradical character and large strain in the
carbon skeleton[4-6]. Therefore C36 cage can be regarded as a
building block to construct 1D, 2D and 3D polymers[6-12]. Some of 1D, 2D and 3D
polymers can be considered as the extension of the rings-structures. A systematic study of
the C36-rings thus is significant in the further study of both C36
solid and the rings-structures itself. In a previous paper we have constructed Dnh
C36-rings from four “bonding belts”
and investigated using self-consistent-field molecular
orbital method (SCF-MO)[13]. However, there are not the four bonding belts only
even for constructing the rings with Dnh symmetry. More C36-ring
structures are necessary to be investigated for understanding their structure-property
relationship. In this paper, the C36-rings are built through another two
bonding belts and studied by the same method as in ref. [13]. The structure and electronic
properties of these C36-rings are discussed and compared with those in the
previous paper[13] and the C60-rings[14].
2. CONSTRUCTION OF C36-RINGS
AND COMPUTATION DETAILS
The C36 cages used to construct the rings here are those with D6h
symmetry and we still focus on the C36-rings with Dnh
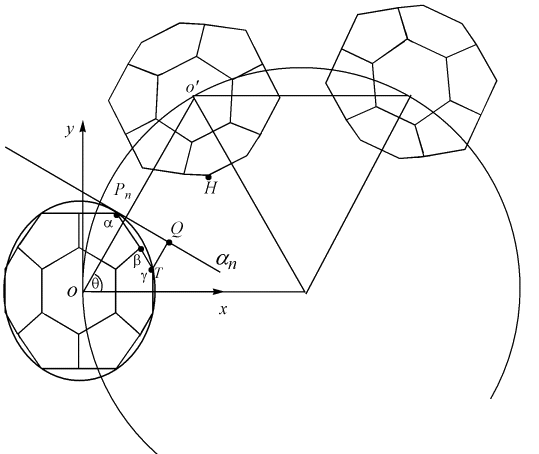
symmetry only. Followed ref. [13] and [14], we build a reference frame in the C36
cage constructing the rings as shown in Fig.1. The center of C36 cage O is
set as the origin point of the reference frame, the x-axis points to the center of the
ring and the z-axis is vertical to the ring plane. Adjacent C36 cages in the
ring will form bonds and the possibly bonding atoms will alter with the change of the ring
size. These possibly bonding atoms are situated in a special area on the surface of the C36
cages, which is defined as a "bonding-belt". We name the two bonding-belts studied here as belt-A and belt-B,
which are shown in Fig.2. It can be seen that the atoms on the two bonding-belts are along
lines from one pole to another pole of the cage. Especially, the bonding belt degenerates
to a line for belt-B. The projections of C36 cages on the ring plane are
ellipses.
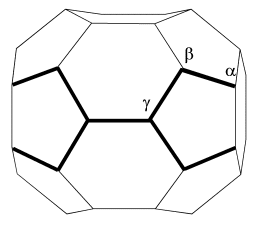
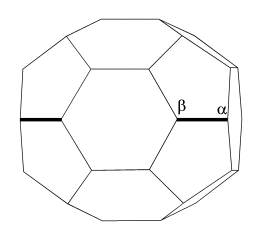
belt A belt B
Fig.2 Two Dnh bonding-belts as shown by bold line area on the C36 cages. The Greek alphabets denote atom indices.
We describe the C36-rings
in the following denotation[13]: <Bonding-belt >-<symmetry (Dnh)>-<linking
pattern described by indices of bonding atom in bonding-belts >. For instance, the ring
A-D3h-aabbg is a
D3h ring composed of three C36 cages and there are five bonds
between two neighboring C36 cages on the Belt-A.
The stabilities and electronic structures of C36-rings
studied in this paper are calculated by semi-empirical SCF-MO method at AM1[15]
level with GAMESS program package[16] and full geometric optimization. The size
of the C36-rings is considered from 3 to 8. Here some of the rings are quite
large, hence the semi-empirical method is still suitable for full geometric optimization.
Besides, a comparison with the previous results can be done based on the same calculation
level. Symmetry constraint on the C36-rings is applied in the calculations, but
the C36 cages can be relaxed from D6h to lower symmetry.
3. RESULTS AND DISCUSSIONS
3.1 Stability
Among the 54 rings produced from the two bonding belts, 19 stable C36-rings
with Dnh symmetry have been obtained by full optimization. The energy
per C36 cage related to that of single C36 with D6h symmetry
(![]() ) is given by:
) is given by:
![]()
where ![]() is the total energy of the C36-rings.
The calculated results of the 19 C36-rings are listed in Table 1.
is the total energy of the C36-rings.
The calculated results of the 19 C36-rings are listed in Table 1.
Table 1. Energy E (Kcal/mol), standard deviation
s (in Å)and intermolecular bond length (Å) of the C36-ringsC36-rings |
E |
s | Bond lengths |
||
Ca-a |
C b-b |
C g-g |
|||
C36 |
0.0 |
- |
- |
- |
- |
A-D4h-aa |
-71.04156 |
0.03113 |
1.530 |
||
A-D5h- aa |
-85.82963 |
0.00293 |
1.521 |
||
A-D6h- aa |
-88.22879 |
0.00069 |
1.520 |
||
A-D7h- aa |
-81.97039 |
0.00689 |
1.517 |
||
A-D8h- aa |
-83.22542 |
0.01557 |
1.523 |
||
A-D3h- aabb |
-125.61846 |
0.11449 |
1.566 |
1.546 |
|
A-D4h- aabb |
-96.03858 |
0.15646 |
1.491 |
1.695 |
|
A-D3h- aabbg |
-123.22106 |
0.10696 |
1.585 |
1.563 |
1.577 |
A-D3h- bb |
-101.80638 |
0.10873 |
1.580 |
||
A-D3h- bbg |
-101.80643 |
0.09417 |
1.596 |
1.562 |
|
A-D4h- bbg |
29.88844 |
0.18630 |
1.617 |
1.643 |
|
B-D5h- a |
-70.94519 |
0.00048 |
1.504 |
||
B-D6h- a |
-69.93688 |
0.01112 |
1.505 |
||
B-D7h- a |
-61.07792 |
0.02694 |
1.512 |
||
B-D8h- a |
-63.85700 |
0.04311 |
1.514 |
||
B-D3h- ab |
-108.60932 |
0.09446 |
1.587 |
1.482 |
|
B-D4h- ab |
-88.94393 |
0.10335 |
1.474 |
1.588 |
|
B-D5h- ab |
-59.25635 |
0.20704 |
1.467 |
2.096 |
|
B-D3h- b |
-103.09204 |
0.01207 |
1.515 |
||
From a view of energy,
theoretical calculations here give that forming rings are favorable to the stabilization
of the systems. This effect is similar to the previous results but quite different from
the situation of the C60-rings, among them more than half become unstable[14].
It can be seen that at least one of the intermolecular bond lengths is
smaller than 1.65
The stability of C36-rings may be affected by several factors in which the change of the strain energy play an important role. Formation of covalent bond between neighbor C36 cages will cause the distortion of the C36 cage, which results in the increase of the strain energy.
The parameter s has been used to estimate the strain change of C36 cage distortion for the rings[13]. We choose D6h C36 cage optimized at AM1 level as a benchmark for the distortion of C36 cage in the rings. As shown in Fig.1 an is the tangent plane of the C36 cage perpendicular to line OO', Pn is the tangent point and T is the position of a possible bonding atom on the bonding-belt. TQ is the vertical distance from the possible bonding atom to an and concerned with the relative distance of possible bonding atom pair, such as T, H. The parameter s then can be obtained by
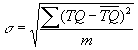
where m is the number of TQ including reference point Pn (TQ =0 at Pn) for a certain linking pattern, such as m=5 for "aabb". The calculation of s is dependent only on the structure of a single undistorted C36 cage, the linking pattern and the ring size. The value of s(Å) for every linking pattern and ring size are listed in Table 2. But only the rings with the values expressed by italic boldface type can be obtained by full optimization. It can be seen that smaller value of s is favorable to the formation of C36-rings. The rings with s bigger than 0.21Å cannot lead to convergent results. From Table 1, it can be inferred that the smaller the the lower the energies of the rings for the same linking pattern except for the two D7h rings. This indicates that the distortion has a great influence upon the stability and that parameter s can be used to determine the order of stability for those rings with the same linking pattern. It should be noted, however, that D7h symmetry is unavailable in the GAMESS program. The two D7h rings are obtained finally by the point-wise optimization. We think this is the main reason why the stability of D7h rings does not follow the order of s.
Table 2. The values of the parameter s for all the rings considered here.
belt |
Linking patterns |
n |
|||||
3 |
4 |
5 |
6 |
7 |
8 |
||
A |
aa |
0.14712 |
0.03113 |
0.00293 |
0.00069 |
0.00689 |
0.01557 |
| aabb | 0.11449 |
0.15646 |
0.25113 |
0.31794 |
0.36632 |
0.40284 |
|
| aabbg | 0.10696 |
0.18954 |
0.31521 |
0.40141 |
0.46302 |
0.50903 |
|
| bb | 0.10873 |
0.16950 |
0.24359 |
0.30640 |
0.35705 |
0.39791 |
|
| bbg | 0.09417 |
0.18630 |
0.28577 |
0.36278 |
0.42180 |
0.46839 |
|
| g | 0.07807 |
0.25230 |
0.39483 |
0.50231 |
0.58407 |
0.64768 |
|
B |
a | 0.10568 |
0.01122 |
0.00048 |
0.01112 |
0.02694 |
0.04311 |
a b |
0.09446 |
0.10335 |
0.20704 |
0.28230 |
0.33859 |
0.38214 |
|
| b | 0.01207 |
0.11480 |
0.21984 |
0.30483 |
0.37184 |
0.42515 |
|
Besides the strain,
other factors such as the type of the bonding atoms, the size of retained conventional
aromatic domains and the double bonds introduced into pentagons have also important
influence on the stability of C36-rings.
The C36 molecule with D6h symmetry has three
kinds of non-equivalent atoms
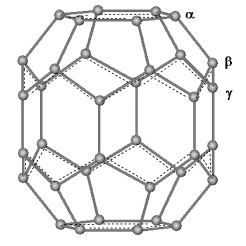
Fig.3 Denotation of three non-equivalent atoms and the aromatic domains (bracketed by dashed lines) on the Dnh C36 cage.
It was pointed out that large
p-orbital axis vector (POAV) angle caused large strain on the sp2 carbon atom and had high reactivity[17]. The sp2 carbon atom with large POAV angle turned into sp3 hybridization will lead to smaller distortion of the carbon cage. The POAV angles are 107.03°, 106.13° and 102.82° respectively for a, b and g. There are no convergent structures for those rings with pure g-g intermolecular bonds here. However, Table 1 shows that the rings through pure a-a connection, such as A-D4h-aaand A-D5h-aa, are less stable than those with pure b-b bonds, such as A-D3h-bb and B-D3h-b. Although a atom has larger POAV angle, formation of a-a bonds destroys the aromaticity of hexagon domains on the polar region of C36 cage. Kroto has proposed that the derivative stability is increased for small fullerenes by maximizing conventional aromatic domains concomitant with alleviating cage strain[18]. The rings containing C36 cages with larger size of conventional aromatic domains should be relatively more stable. In addition, if a double bond is produced on the shared side of two abutting pentagons, larger strain will be introduced into the C36 cage and decrease the stability of the rings. The shared pentagon-pentagon bonds of isolated D6h C36 cage can be considered as single bonds (1.511Å calculated by AM1). It is found that the stability of rings increase as the pentagon-pentagon bond tends to be a single bond. For instance the shared pentagon-pentagon bonds of the most stable ring (A-D3h-aabb) are all longer than 1.511Å while the unstable ring (A-D4h-bbg) has four of the bonds with the lengths of 1.409Å.3.2 Electronic structures.
The calculated energy levels and symmetries of the higher occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), as well as the energy gaps between them (Eg.), are listed in Table 3 for the C36-rings. It can be seen that the energy gaps are all bigger than that of C36 cage except A-D4h-aa. Here the most stable ring (A-D3h-aabb) has the largest energy gap with the value of 5.92ev. It was reported that the energy gaps are reverent to kinetic stability of the fullerene[19], larger energy gap results in high kinetic stability. Thus forming of ring structures is favorable to both kinetic and thermodynamic stability of the systems. The rings formed through b-b bonds have relatively large Eg such as A-D3h-bb and B-D3h-b while pure a-a bond leads to comparatively small ones such as A-D4h-aa and A-D5h-aa.
It can also be found that doubly degenerated frontier orbitals (HOMOs or LUMOs) occur mainly in the rings with odd number of C36 cages. For the rings with even size (n=2,4,6,8), only HOMO of the ring A-D4h-bbg is degenerated with Eg symmetry. This is some similar to the cases of the C60-rings, the doubly degenerated frontier orbitals are produced only for those with odd size (n=3,5,7)[14]. Different distribution of energy levels will give different character of spectrum, which may help us to determine the ring structures.
Table 3. Energy gaps (Eg), HOMO and LUMO levels and symmetry of Dnh C36-rings (in ev)
C36rings |
Eg |
HOMO |
LUMO |
||
Single C36 |
3.08312 |
-7.87243 |
B1u |
-4.78931 |
B2g |
A-D4h- aa |
2.70215 |
-7.76358 |
B2u |
-5.06143 |
B1u |
A-D5h- aa |
3.62736 |
-8.32687 |
E2'' |
-4.69951 |
E2'' |
A-D6h- aa |
3.85866 |
-8.48198 |
B2g |
-4.62332 |
A1u |
A-D7h- aa |
3.17015 |
-8.00565 |
E2' |
-4.83550 |
E2' |
A-D8h- aa |
4.01649 |
-8.58811 |
B2u |
-4.57162 |
A2u |
A-D3h- aabb |
5.91588 |
-9.19493 |
E'' |
-3.27905 |
E'' |
A-D4h- aabb |
5.57846 |
-9.06160 |
B1u |
-3.48314 |
B2u |
A-D3h- aabbg |
5.87507 |
-9.19221 |
E'' |
-3.31714 |
E'' |
A-D3h- bbg |
5.67643 |
-9.24664 |
E'' |
-3.57021 |
A2'' |
A-D4h- bbg |
4.79203 |
-9.14051 |
Eg |
-4.34848 |
B2g |
A-D3h- bb |
5.70363 |
-9.25480 |
A2' |
-3.55117 |
A2'' |
B-D5h- a |
5.03966 |
-8.86567 |
A2' |
-3.82601 |
A2'' |
B-D6h- a |
5.03422 |
-8.86839 |
A2g |
-3.83417 |
A2u |
B-D7h- a |
4.61509 |
-8.74581 |
E1'' |
-4.13072 |
E2'' |
B-D8h- a |
5.00156 |
-8.84662 |
B1g |
-3.84506 |
B1u |
B-D3h- ab |
5.55927 |
-9.22201 |
A1' | -3.56200 |
E' |
B-D4h- ab |
4.76755 |
-8.56362 |
A1g |
-3.79607 |
A2g |
B-D5h- ab |
5.58390 |
-9.33099 |
E1' |
-3.74709 |
E2' |
B-D3h- b |
5.37164 |
-9.03438 |
A2' |
-3.66274 |
A1' |
4. CONCLUSION
The Dnh C36-rings constructed through the two new
bonding-belts are investigated using the semi-empirical AM1 molecular orbital method. The
strain-associated factor
REFERENCE
[1] Piskoti C, Yarger J, Zettl A. Nature, 1998, 393: 771-774.
[2] Koshio A, Inakuma M, Zhong W et al. J. Phys. Chem., 2000, 104: 908-7913.
[3] Koshio A, Inakuma M, Sugai T et al. J. Am. Chem. Soc., 2000, 122: 398-399.
[4] Yuan L, Yang J, Deng K et al. J. Phy. Chem., A 2000, 104: 6666-6671.
[5] Ito A, Monobe T, Yoshii T et al. Chem. Phys. Lett., 2000, 328: 32-38.
[6] Jagadeesh MN, Chandrasekhar J. Chem. Phys. Lett, 1999, 305: 298-302.
[7] Chen Y, Li Y, Huang Y et al. Acta Chimica Sinica, 2000,58 (12): 1511-1515.
[8] Huang Y, Chen Y, Liu R. J. Phys. Chem. Solids, 2000, 61: 1475-1481.
[9] Fowler PW, Heine T, Rogers KM et al. Chem. Phys. Lett., 1999, 300: 369-378.
[10] Menon M, Richter E. Phys. Rev., B 1999, 60 (19): 13322-13324.
[11] Grossman JC, Louie SG, Cohen ML. Phys. Rev., B 1999, 60 (10): 6941-6944.
[12] Heine T, Folwer PW, Seifert G. Solid State Commun., 1999, 111: 19-22.
[13] Li Y, Huang Y, Du S et al. Carbon, 2002, 40: 2255-2262.
[14] Li Y, Huang Y, Du S et al. Chem. Phys. Lett., 2001, 335: 524-532.
[15] Dewar MJS, Zoebisch EG, Healy EF. J. Am. Chem. Soc., 1985, 107: 3902-3909.
[16] Schmidt MW, Baldridge KK, Boatz JA et al. J. Comput. Chem., 1993, 14: 1347.
[17] Haddon RC. Science, 1993, 261: 1545-1550.
[18] Kroto HW, Walton DRM. Chem. Phys. Lett. 1993, 214: 353-356.
[19] Manolopoulos DE, May JC, Down SE. Chem. Phys. Lett., 1991, 181: 105.