http://www.chemistrymag.org/cji/2004/06c087nc.htm |
Dec. 1,
2004 Vol.6 No.12 P.87 Copyright |
Preparation, biological activity and quantum chemistry calculation of quercetin-Aluminium complex
Jiang
Liuyun, Liu Yuming
(College of Chemistry, Sichuan University, Chengdu 610064)
Keywords quercetin, complex, biological activity, ab initio calculation 槲皮素-铝配合物的合成、生物活性及其结构的量子化学计算
蒋柳云 刘玉明
(四川大学化学学院 成都 610064)
2004
年9月22日收稿 摘要 从溶液中合成了槲皮素-铝配合物,通过元素分析、红外光谱、核磁共振、差热分析确定其组成为Al(C15H9O7)3·4H2O;并用邻苯三酚自氧化法测定得出其有较强的抗氧化活性;最后在HF/LanL2DZ基组水平上对其结构进行了量子化学计算,讨论了其几何构型、振动频率、电荷布居及分子轨道,从而进一步从理论上给出了该配合物的能稳定存在及有较强的抗氧化活性的原因,使理论与实验相统一。关键词 槲皮素, 配合物, 生物活性, 从头计算
铝是人体内的一种微量元素,正常总含量约为45-150mg[1]。由于含铝制品应用广泛,人体中铝的含量有逐渐升高的趋势。文献报道许多疾病与体内铝含量过高有关[2, 3]。铝毒性的分子学机理研究表明,铝的有毒形态是自由态Al3+。因此,能够与自由态Al3+形成稳定配合物的配体可降低自由态Al3+在体内的浓度,从而降低或解除铝对人体的毒性[4]。槲皮素(3, 5, 7, 3',4'五羟基黄酮,quercetin)是一种具有抗炎、抗病毒及抗氧化等作用的黄酮醇类化合物[5],近年来,人们发现槲皮素与Cu、Zn、Ni等大部分过渡金属离子及稀土离子都能形成具有较强的清除超氧阴离子自由基能力的配合物[6],还可降低毒副作用。对于槲皮素与主族元素铝形成配合物的研究,J.P.Cornard 等人[7]通过考查不同pH值下AlCl3与槲皮素紫外光谱,借助于溶液体系的谱学数据,对配合物的组成与结构进行了推测,但对于该配合物的结构及抗氧化活性的研究,都未能给出进一步研究。基于此,本文以槲皮素为配体,从溶液中合成得到了槲皮素-铝配合物,并对其进行了生物活性测试,最后对其结构在HF/LanL2DZ基组水平上进行了量子化学计算,从而进一步从理论上给出了该配合物的结构及有较强的抗氧化活性的原因,为槲皮素可作为体内过量铝的解毒剂提供了重要依据,而且为开发抗氧化性槲皮素配合物提供了一定的参考价值。 1 实验部分
1.1 试剂和仪器
所有化学试剂均为分析纯,C、H元素分析用Perkin-Elmer 2400元素分析仪,铝含量用PLASMA-SPECI电感偶合等离子体发射光谱仪,红外光谱用日本岛津FTIR-8700红外光谱仪(4000-400cm-1, KBr压片),差热分析用杜邦1090-B差热分析仪,核磁共振用FT-80A核磁共振仪,生物活性检测用751型紫外可见分光光度计完成。
1.2 配合物的合成
在带有搅拌器、干燥管、冷凝管的150ml的三口瓶中加入槲皮素0.9mmol(约0.3042g)和无水乙醇,加热搅拌使槲皮素溶解,再加入适量乙醇钠溶液,反应一段时间后,加入0.3mmol(约0.0725g) AlCl3·6H2O,然后加热回流搅拌,产生淡黄色沉淀,冷却、抽虑,用乙醇-水洗涤,再用水洗多次,固体产物在真空中干燥。
1.3 生物活性测试
采用化学方法----邻苯三酚自氧化法测定了槲皮素及其配合物对超氧阴离子自由基的清除率。
1.4 量子化学计算
利用HyperChem软件包构建分子模型,先用分子力学MM+进行初步优化,然后利用Guassian98程序[8], 依次在HF/3-21G、HF/6-31G*、HF/LANL2DZ基组水平上对其进行全优化从头计算,计算模型如图1所示。该计算共涉及94个原子,656个基本函数,1770 个初始高斯函数,其中234个为占据轨道。全部计算在PentiumIV上进行。
2
结果与讨论2.1 配合物组成确定
2.1.1 元素分析
配合物元素分析结果(%)为:计算值:C, 53.89;H, 3.49;Al, 2.69;实验值:C, 53.27;H, 3.23;Al, 2.63.
2.1.2 红外光谱实验
表1列出了槲皮素及其配合物红外光谱数据及其归属。从表1可知,槲皮素中羰基振动频率位于1664cm-1,形成配合物后,位移至1632cm-1,而其它键的振动频率基本保持不变,可见配体的羰基氧参与了配位。且在682 cm-1处有新的吸收峰出现,则证明金属离子已参与了配位。
表
1 槲皮素及其配合物的主要红外光谱数据及归属Table 1 IR data of the quercetin and its complex
| 化合物 | n(O-H) | n(C=O) | n(C=C) | n(C-OH) | n(C-O-C) | n(Al-O) |
| 槲皮素 | 3409-3144 | 1664 | 1612 | 1382 | 1262 | - |
| 配合物 | 3455-3136 | 1632 | 1604 | 1372 | 1266 | 682 |
2.1.3 核磁共振实验
为了进一步探讨槲皮素与金属的配位情况,应用FT-80A核磁共振仪,以d6-丙酮为溶剂,测定了槲皮素及其配合物的1HNMR谱,槲皮素归属为:d 12.50(1H,5-OH);d 10.81(1H,7-OH);d 9.57(1H,3-OH);d 9.32(1H,4'-OH);d 9.27(1H,3'-OH);d 7.66(1H,H-2');d 6.90(1H,H-6');d 6.53(1H,H-5');d 6.85(1H,H-8);d 6.17(1H,H-6);d 3.3-4.0(宽峰,H2O)。 配合物归属为:d 11.82(1H,5-OH);d 10.83(1H,7-OH);d 9.26(1H,4'-OH);d 9.19(1H,3'-OH);d 7.81(1H,H-2');d 7.16 (1H,H-6');d 6.57(1H,H-5');d 6.48(1H,H-8);d 6.18(1H,H-6);d 3.0-3.6(宽峰,H2O)。
比较槲皮素与配合物的1HNMR谱,发现槲皮素于d9.57(1H,3-OH)的吸收峰在配合物中消失,表明配位时3-OH失去质子并通过氧原子与金属离子配位。
2.1.4 差热分析
配合物的热谱分析表明,配合物在低于1000C时有一失水吸热峰,失重率约7.25%,与4个水分子的量相对应,在高于1000C时,没有失重现象,说明该配合物含4个结晶水,不含配位水。
综合以上表征结果,得该配合物的分子式为Al(C15H9O7)3·4H2O,中心结构如图1所示。
2.2 生物活性测定
该配合物微溶于水,难溶于CCl4,可溶于甲醇、乙醇、丙酮,易溶于甲酸、DMSO等。把槲皮素及其配合物分别溶于甲醇,采用化学方法----邻苯三酚自氧化法[9]测定了它们在不同浓度下对超氧阴离子自由基的清除率(%),测定结果见表2。由表2数据可知,配合物对超氧阴离子自由基(O2-。)的清除率比槲皮素在不同浓度下都有提高,且清除率不一定与浓度成正比变化。
表
2 槲皮素及其配合物对超氧阴离子自由基的清除率Table 2 Date of scavenging effects on O2·- radical of quercetin and its complex
| 化合物 | 对超氧阴离子的抑制率/% | |||||
| 100 | 300 | 500 | 1000 | 1500 | 2000 (mg/10ml) | |
| 槲皮素 | 41.33 | 63.90 | 83.90 | 39.81 | 65.44 | 72.48 |
| 配合物 | 45.43 | 72.57 | 85.05 | 53.90 | 73.33 | 74.48 |
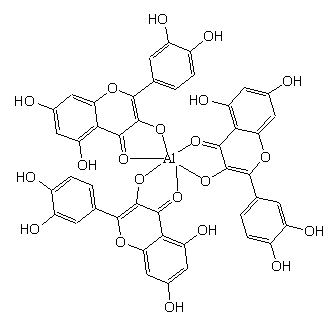 |
 |
| 图1
槲皮素-铝的结构示意图 Fig.1 Structure of quercetin-Aluminium complex |
图2 槲皮素-铝配合物的优化结构 Fig.2 Optimized structure of quercetin-Aluminium complex |
2.3 量化计算结果
2.3.1 几何构型讨论
图2是最后优化得到的槲皮素-铝配合物结构,表3列出了该配合物优化结构的部分参数。从表3数据可知,该配合物结构优化后,对称性好。从分子结构图可见,分子中每个槲皮素的羰基和3-OH同时与Al3+配位,3个配体中的6个键合氧原子形成六配位八面体配合物,由于槲皮素与Al3+键合的2个氧原子分别来自羟基和羰基,致使所形成的Al-O键长不等,Al-O键的平均距离为0.19355nm,再比较Al3+与其配位的氧原子形成的配位键长,发现Al3+与羰基氧形成的键长比与羟基氧形成的键长略长,说明其与羰基氧形成的配位作用略弱;平均键角<O-Al-O为90.14960C (两氧相邻)、169.8290C (两氧不相邻),即形成扭曲的六配位八面体结构。分子中每个Al3+与槲皮素形成3个螯合环,均为非等边五元环,使配合物呈“风车型”结构。对于每个槲皮素配体,从二面角可看出,每一个槲皮素配体的A、C环上所有原子几乎共平面,B环上所有原子也几乎共平面,但B环与A、C平面不完全共平面,成约2-30C的角度;从键长可知,所有C-C单双键键长出现平均化,C-O单键键长都小于正常的C-O单键的键长(0.143nm),C=O双键也大于正常的C=O双键的键长(0.122nm),说明氧原子也参与了共轭,即每个槲皮素配体可能是通过一个共轭大p键形成的近似的平面结构,其中B环与A、C环共轭性略差。
表3 槲皮素-Al配合物的优化结构的结构参数
Table 3 Optimized
structure parameters of quercetin- Aluminium complex
键 键长(nm) |
键 键长(nm) |
键 键长(nm) |
键 键长(nm) |
键 键长(nm) |
||
C34-C35 0.14664 |
C49-C58 0.14018 |
C52-O62 0.13572 |
C35-O25 0.13332 |
Al21-O26 0.19932 |
||
C35-C42 0.13505 |
C34-C41 0.14393 |
C73-O86 0.13673 |
Al21-O28 0.19875 |
Al21-O27 0.18754 |
||
O50-C42 0.13945 |
C41-C51 0.14053 |
C70-O83 0.13941 |
Al21-O25 0.18753 |
|||
C42-C49 0.14632 |
C51-C61 0.13946 |
C82-O89 0.13735 |
Al21-O16 0.20173 |
|||
C49-C59 0.14048 |
C41-C52 0.14204 |
C34-O28 0.12607 |
Al21-O20 0.18644 |
|||
键角 (0C) |
键角 (0C) |
键角 (0C) |
键角 (0C) |
|||
O25-Al21-O28 80.1912 |
O25-Al21-O26 90.6944 |
O20-Al21-O28 94.8753 |
O25-Al21-O20 168.2704 |
|||
O16-Al21-O27 170.641 |
O26-Al21-O20 94.5246 |
O28-Al21-O26 170.5756 |
O25-Al21-O16 90.0892 |
|||
O16-Al21-O20 79.2538 |
O27-Al21-O26 81.6261 |
O27-Al21-O28 96.9719 |
O28-Al21-O16 90.4396 |
|||
O16-Al21-O26 91.9779 |
O27-Al21-O20 94.3714 |
O27-Al21-O25 96.7798 |
||||
二面角 (0C) |
二面角 (0C) |
二面角 (0C) |
二面角 (0C) |
|||
O86-C73-C60-C52 179.9891 |
O62-C52-C41-C51 -179.556 |
O28-C34-C35-C42 -179.9292 |
H84-C69-C58-C49 179.8566 |
|||
O25-C35-C34-C41 -179.9368 |
O83-C70-C59-C49 179.9823 |
O89-C82-C69-C58 -179.9624 |
H78-C60-C52-C41 -179.9956 |
|||
C59-C49-C42-C35 177.17 |
C58-C49-C42-C50 176.8913 |
C61-C51-C41-C34 179.6743 |
H72-C59-C49-C58 -179.8753 |
|||
O50-C51-C41-C52 179.6173 |
H92-O86-C73-C60 179.9937 |
H77-O62-C52-C41 -179.9106 |
H71-C58-C49-C59 179.9193 |
|||
H90-O83-C70-C59 -2.9176 |
H93-O89-C82-C69 179.7647 |
H76-C61-C51-C41 -179.9742 |
||||
2.2.2
振动频率讨论为了进一步确定槲皮素-铝配合物能稳定存在,本文对在HF/LanL2DZ水平优化得到的平衡构型进行了振动频率计算。由计算结果可知,该构型共计算了276个频率,所有振动频率均为正值,其中最小的振动频率为5.2260 cm-1,强度为0.1430 km/mole。说明没有虚频,从而保证了其能量的二阶导数矩阵的本征值为正值,则相应的构型应为稳定结构[10]。
2.3.3 电荷布居讨论
由Mulliken布居分析, 得到原子净电荷布居。表4列出了Al及部分原子的净电荷布居。从表4可知,参与配位的氧原子净电荷均为负,形成一负电荷的空穴,Al3+处于空穴的中心,Al3+的净电荷为1.805,而不是原来的(+3),说明Al和O原子电子云相互交叠,形成了配位键。且由其净电荷可以预见,Al3+仍具有成键能力,即具有接受O2·- 的孤对电子的能力,因而具有较高的抗氧化活性[11]。
表4 在HF/LanL2DZ水平下槲皮素-Al配合物的部分原子净电荷
Table 4 Net charges of some atoms of quercetin-Aluminium complex at HF/LanL2DZ level
原子 电量 |
原子 电量 |
原子 电量 |
原子 电量 |
原子 电量 |
原子 电量 |
原子 电量 |
O27 -0.839 |
O62 -0.572 |
O7 -0.574 |
O81 -0.638 |
C14 0.285 |
C6 0.000 |
C15 0.284 |
O26 -0.739 |
O86 -0.608 |
O8 -0.612 |
O88 -0.668 |
C35 0.271 |
C41 -0.004 |
C49 0.270 |
O25 -0.845 |
O83 -0.638 |
O17 -0.572 |
Al21 1.805 |
C33 0.305 |
C38 -0.882 |
C48 0.271 |
O28 -0.589 |
O89 -0.669 |
O55 -0.625 |
C10 0.456 |
C11 0.254 |
C3 0.457 |
|
O16 -0.602 |
O40 -0.625 |
O79 -0.608 |
C34 0.447 |
C42 0.264 |
C51 0.453 |
|
O20 -0.837 |
O36 -0.680 |
O45 -0.564 |
C32 0.485 |
C39 0.267 |
C44 0.448 |
2.3.4
分子轨道讨论为进一步探求配合物的活性部位的特征及成键规律,对配合物的分子轨道进行了分析,用参与组合的各类原子轨道系数的平方和来表示该类原子轨道对分子轨道的贡献,并经归一化后得到结果。表5列出了前沿分子轨道能量及原子轨道对前沿分子轨道的贡献。从表5数据可以看出:从分子轨道的能量来看,其占据的分子轨道能量都较负,表明配合物的电子状态是稳定的,其光电子能谱是有意义的[12];分子轨道的能量差值ΔE较大,说明分子能稳定存在[13]。从前沿分子轨道的成分和所占的比重来看,在5条占有轨道中,轨道成分较分散,它们被分配到形成配合物的各个原子上,说明配合物整个电子云分布在组成分子的各个原子上,且相互之间结合紧密;在最低空轨道及附近的空轨道中,Al的贡献基本上比其它原子的贡献大,可见Al作为其活性中心,有较强的接受O2-·孤对电子的能力,因而具有较高的抗氧化活性[14]。还可看出,在占有轨道中,O62比O25贡献小,O17比O20贡献小,O55比O27贡献小,故很难给出电子,即很难与金属形成配位键[15],故槲皮素在与Al3+配位时,是3-OH参与配位,而不是5-OH参与配位。还发现在占有轨道中羰基氧的贡献比羟基氧基本上要小,即提供电子的能力要弱,故形成的配位键要弱,这与前面对键长和电荷布居的讨论结论相一致。总之,通过对分子轨道的讨论,进一步认识了它们的成键特性及有催化超氧阴离子能力的抗氧化活性的本质。 表5 在HF/LanL2DZ水平下槲皮素-Al配合物的前沿分子轨道能量及原子轨道对前沿分子轨道的贡献(%)
Table 5 Some frontier molecular orbital energies(eV)and atomic orbitals contribution to molecular orbitals (%) of quercetin-Aluminium complex at HF/LanL2DZ level
分子轨道 |
H-4 |
H-3 |
H-2 |
H-1 |
HOMO |
LUMO |
L+1 |
L+2 |
L+3 |
L+4 |
能量 (eV) |
-8.84952 |
-8.741536 |
-7.672848 |
-7.570576 |
-7.52216 |
1.01592 |
1.101056 |
1.198704 |
2.751008 |
2.849472 |
Al21 |
0.0657 |
0.1256 |
1.3478 |
1.5779 |
0.5996 |
18.575 |
21.620 |
18.780 |
7.019 |
6.265 |
O25 |
0.1081 |
- |
1.1125 |
0.8375 |
11.2853 |
0.0520 |
0.2430 |
0.0309 |
0.7515 |
- |
O28 |
- |
0.0500 |
0.2488 |
0.2620 |
2.3676 |
0.9294 |
- |
0.0810 |
- |
0.0197 |
O16 |
- |
0.2277 |
0.0199 |
2.3667 |
0.4202 |
0.0550 |
0.1688 |
1.0169 |
- |
0.0867 |
O20 |
0.0506 |
4.0427 |
0.1699 |
1.8146 |
1.0532 |
0.0987 |
0.0136 |
0.2570 |
- |
0.6964 |
O26 |
0.1632 |
0.0866 |
1.7811 |
0.1781 |
0.4246 |
0.2998 |
0.0609 |
- |
0.0209 |
0.0425 |
O27 |
3.6176 |
- |
11.1043 |
0.0561 |
2.7084 |
- |
0.9904 |
0.2317 |
0.0594 |
- |
O62 |
- |
- |
0.0650 |
0.0438 |
0.4343 |
- |
1.7587 |
0.0188 |
0.1710 |
- |
O17 |
- |
0.6311 |
- |
0.6230 |
0.0481 |
- |
0.0262 |
1.9392 |
- |
0.1320 |
O55 |
2.21 |
- |
0.9810 |
- |
0.1308 |
1.6600 |
- |
- |
- |
- |
本文从溶液中合成了槲皮素-铝配合物,通过元素分析、红外光谱、核磁共振、差热分析确定其组成为Al(C15H9O7)3·4H2O,并通过邻苯三酚自氧化法测定得出其有较强的抗氧化活性,最后在HF/LanL2DZ基组水平上进行了量子化学计算,讨论了其几何构型、振动频率、电荷布居及分子轨道,从而进一步从理论上认识了该配合物的稳定结构及有较强的抗氧化活性的原因,使理论与实验相统一。该研究不仅为槲皮素可作为体内过量铝的解毒剂提供了重要依据,而且为开发抗氧化性槲皮素配合物提供了一定的参考价值。
REFERENCES
[1] Peng A, Wang W H. Environ, Bio-inorganic Chemistry, Beijing, Beijing University Press,
1991: 151-155.
[2] Micheal R W. Environ Health Perspectives, 1985, 63: 141.
[3] Macdonald T L, Martin R B. Trend, Biochem. Sci., 1988, 13:15-21.
[4]Wu Y Q, Zhang B, Zhu, Q T et al. Chen J. Chem J Chin Univ (Gaodengxuexiao huaxue
xuebao).1998,19 (3): 410-413.
[5] Ji Y B, Pharmacology and applied of effective ingredient from Chinese traditional
medicine. Science Press of Heilongjiang, Haerbin, 1994: 261-263.
[6] Zhou J, Wang L F, Wang J Y et al. Journal of Inorganic Biochemistry . 2001, 83: 41-48.
[7] Cornard J P, Merlin J C. Journal of Inorganic Biochemistry . 2002, 92: 19-27.
[8] Aeleen F , Michael J F. Gaussian 98 User's Reference, Gaussian , Inc., Garnegie Office
Park , Bldg. 6 Pittaburgb , PA15106 USA.
[9] Fan Z, Zhao R, Zhou W H et al . J. of Tianjin Normal University (Natural Sciences
Edition)(Tianjin shifan daxue xuebao). 1999, 19 (3): 16-21.
[10] He W D, Zhou G, Hu, H R et al. Acta Chimica Sinica
(Huaxue xuebao), 2001,59 (8): 1210-1215.
[11] Chen K,Yin Y X, Sun Y et al. J. of Tianjin Normal University(Natural
Sciences Edition)(Tianjin shifan daxue xuebao), 2003, 23 (1): 4-7.
[12] Zheng K C, Huang J D, Shen Y et al. Chinese
J.of Inorganic Chem (Wuji huaxue xuebao). 1999,15 (1): 74-77.
[13] Zhang F X, Kuang D Z, Xu Z F et al. Chinese J. of Inorganic Chem (Wuji huaxue xuebao).2002,
18 (8): 854-858.
[14] Liu X H, Liu X L, Yue J J et al. Chinese J. Struct. Chem (Jiegou huaxue). 2004, 23
(3): 312-315.
[15] Liu X L, Zhao R, Liu X H et al. Acta Chimica Sinica (Huaxue xuebao). 2003, 61 (4):
578-583.