http://www.chemistrymag.org/cji/2005/077049ne.htm |
Jul. 2, 2005 Vol.7 No.7 P.49 Copyright |
A facile procedure for synthesis of N-sulfonylimines catalyzed by silica sulfate solid acid
Geng Lijun, Liu Yan
(Department of Chemistry, Handan College,Handan,056005)
Received on Apr.18, 2005.
Abstract N-sulfonylimines have been successfully synthesized by the reaction of aldehydes with sulfonamides in the presence of SiO2-OSO3H solid acid. This method is fast and excellent yielding for a range of substrates.Keywords N-sulfonylimines, aromatic aldehydes, sulfonamides
Recently, the synthesis and application of
N-sulfonylimines from aldehydes with sulfonamides has been found to be very useful in
organic chemistry. N-sulfonylimines are powerful synthetic intermediates[1,2]
in organic synthesis and industrial application. They are used in numerous reactions such
as inverse electron-demand Diels-Alder reactions,[3,4] addition
reactions as carbonyl equivalents[5] and ene reactions.[6] Several
synthetic methods for the preparation of N-sulfonylimines have be reported in the
literature.[7-14] Some of these methods have not been entirely satisfactory
owing to such drawbacks as long reaction times, expensive and hazardous reagents,
non-recoverable catalysts, cumbersome experimental and requiring use of a microwave oven.
Solid acid is an efficient and facile catalyst for a variety of organic
reactions. [15-16] Herein we describe a facile and efficient method for the
synthesis of N-sulfonylimines from aldehydes with sulfonamides catalyzed by SiO2-OSO3H
solid acid (Scheme 1).
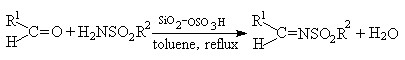
1
2
3
Representative results
that we have obtained for the preparation of N-sulfonylimines are summarized in Table 1.
It was found that aromatic and aliphatic aldehydes in the presence of SiO2 -OSO3H
are heated with sulfonamide in refluxing toluene to offer the corresponding
N-sulfonylimines in good yield. The reaction proceeds cleanly and work-up is simple. The
product can be obtained by only filtration of the catalyst and removing solvent.
In the absence of catalyst, lower yields of product are observed even
with prolonged reaction time. For example, entry 2 without catalyst after 2 hours only 40%
yield of product is obtained in refluxing toluene, whereas 91% yield is obtained with
catalyst for 0.5h. The reaction worked better in refluxing toluene than in refluxing
benzene.
Table 1 Conversion of aldehydes with sulfonamides into N-sulfonylimines in the presence of SiO2-OSO3H
Entry |
R1 |
R2 |
Product |
Time |
Yieldsa |
Mp/ ºC |
|
Found |
Reported |
||||||
1 |
C6H5 |
4-CH3C6H4 |
3a |
40 |
95 |
113-114 |
11013 |
2 |
C6H5 |
C6H5 |
3b |
30 |
91 |
140 |
139-1419 |
3 |
4-CH3C6H4 |
4-CH3C6H4 |
3c |
65 |
92 |
116-117 |
116-11817 |
4 |
4-CH3C6H4 |
C6H5 |
3d |
60 |
89 |
136-138 |
13617 |
5 |
2-HOC6H4 |
4-CH3C6H4 |
3e |
60 |
91 |
125 |
12517 |
6 |
2-HOC6H4 |
C6H5 |
3f |
75 |
85 |
119-121 |
119-12017 |
7 |
4-CH3OC6H4 |
4-CH3C6H4 |
3g |
55 |
96 |
127-128 |
128-12912 |
8 |
4-CH3OC6H4 |
C6H5 |
3h |
65 |
90 |
146-147 |
147-1499 |
9 |
3,4-(OCH2O)C6H3 |
4-CH3C6H4 |
3i |
35 |
94 |
118-120 |
114-11612 |
10 |
4-ClC6H4 |
4-CH3C6H4 |
3j |
70 |
72 |
173-175 |
172-17317 |
11 |
4-ClC6H4 |
C6H5 |
3k |
75 |
67 |
128-130 |
127-1309 |
12 |
3-ClC6H4 |
C6H5 |
3l |
65 |
75 |
110-112102-103 |
102-10418 |
13 |
3-NO2C6H4 |
4-CH3C6H4 |
3m |
80 |
60 |
142-144 |
143-1459 |
14 |
2,4-Cl2C6H3 |
4-CH3C6H4 |
3n |
70 |
71 |
111-113 |
112-11317 |
15 |
C6H5CH=CH |
4-CH3C6H4 |
3o |
35 |
96 |
109-110 |
108-1091 |
16 |
C6H5CH=CH |
C6H5 |
3p |
30 |
93 |
110-112 |
112-11317 |
a
Yield of pure isolated products. The results show that the
aldehydes with eletron-withdrawing group (1j-1n) need longer reaction time than the
aldehydes with donate group (1c-1i, 1o). This indicates that
electron-donating groups have increased the reaction speed as well as reaction yields. On
the other hand electron-withdrawing groups have decreased yields and increased reaction
time. We have studied the reaction of sulfonamide with ketones and found that the yield is
lower than the reaction of sulfonamide with aldehydes. For example, the yield of
acetophenone treated with p-CH3C6H4SO2NH2
is 42% after refluxing 100 min and there is no product for the reaction of diphenyl ketone
with p-CH3C6H4SO2NH2.
Finally, the conversion rate of p-toluenesulfonamide with aldehydes is a little
higher than benzene sulfonamide.
The catalyst is easily regenerated by washing with ethanol followed by
drying at 100ºC for 2h. The
catalyst could be reused six times for the synthesis N-(p-toluenesulfonyl)benzaldimine
(3a) without significant loss of activity.
In conclusion, SiO2-OSO3H as solid acid catalyst
can offer definite advantages over traditional catalysts in terms of operational
simplicity, short reaction time, non-polluting and high yields for synthesis of
N-sulfonylimines by the reaction of arylsulfonamide and arylaldehydes.
Melting points were determined with a digital electrothermal apparatus
without further correction. IR spectra were recorded on a BIO-RAD FTS-40 IR spectrometer
using KBr pellets. 1H NMR spectra were determined on a Varian VXP-400s
spectrometer using CDCl3 as solvent and tetramethylsilane (TMS) as internal
reference. The products were also characterized by comparison of their melting point with
the literature values. SiO2-OSO3H was obtained as follows: a series
of different concentration of aqueous H2SO4 were prepared firstly,
blending SiO2 with the same volume of aqueous H2SO4 in
vacuum and at room temperature the mixture storing in a desiccator for 12 to 15 hours. The
mixture was stirring at 70-80ºC until it was dried. After then drying them at 120ºC for
4 to 5 hours and the white powder of SiO2-OSO3H was obtained
finally.
A mixture of aldehyde (2.00mmol), sulfonamide (2.00mmol), SiO2-OSO3H
(100mg) and toluene (5ml) was stirred under refluxing for 30-80 mins (Table 1) in a
Dean-Stark apparatus. The reaction was monitored by TLC. After completion of the reaction,
the reaction mixture was cooled to room temperature and SiO2-OSO3H
was filtered off and washed with toluene. The solvent was evaporated under reduced
pressure to provide crude product and the crude product was purified by recrystallization
with hexane-ethyl acetate mixtures or by flash chromatography on silica gel (Et2O-hexane
as eluent) to give the pure product.
The carbon atoms of arylring are numbered as follows:
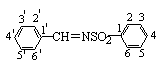
Spectroscopic data of some of the
products are given below:
3a: IR (KBr): nmax 3006, 2948, 1650, 1600, 1578, 1456, 1354, 1162, 812,
750, 680 cm-1; 1H NMR (CDCl3): d H 9.03 (1H, s, HC=N), 7.91 (2H, d, J=7.6 Hz,
2',6'-ArH), 7.88 (2H, d, J=8.0 Hz, 2,6-SO2ArH), 7.60 (1H, t, J=7.6 Hz, 4'-ArH),
7.46 (2H, t, J=7.6 Hz, 3',5'-ArH), 7.35 (2H, d, J=8.0 Hz, 3,5-SO2ArH), 2.45
(3H, s, CH3).
3c: IR (KBr): nmax
3012, 2918, 1652, 1594, 1580, 1490, 1328, 1164, 848, 824 cm-1; 1H NMR (CDCl3): d H 9.02 (1H, s, HC=N), 7.89 (2H, d,
J=8.0 Hz, 2,6-SO2ArH), 7.82 (2H, d, J=7.6 Hz, 2',6'-ArH), 7.36 (2H, d, J=8.0 Hz,
3,5-SO2ArH), 7.30 (2H, d, J=7.6 Hz, 3',5'-ArH), 2.44 (6H, s, CH3).
3e: IR (KBr): nmax
3360, 3044, 2920, 1664, 1616, 1600, 1554, 1296, 1170, 1096, 806, 742 cm-1; 1H NMR (CDCl3):
d H 10.82 (1H, s, -OH),
9.12 (1H, s, HC=N), 7.88 (2H, d, J=8.0 Hz, 2,6-SO2ArH), 7.50 (2H, m,
4',6'-ArH), 7.35 (2H, d, J=8.0 Hz, 3,5-SO2ArH), 7.00 (2H, m, 3',5'-ArH), 2.46
(3H, s, CH3).
3j: IR (KBr): nmax
3028, 2930, 1660, 1604, 1554, 1500, 1330, 1160, 832, 800 cm-1; 1H NMR (CDCl3): d H 9.03 (1H, s, HC=N), 7.88 (2H, d,
J=7.6 Hz, 2,6-SO2ArH), 7.89 (2H, d, J=8.0 Hz, 2',6'-ArH), 7.45 (2H, d, J=8.0 Hz,
3',5'-ArH), 7.34 (2H, d, J=7.6 Hz, 3,5-SO2ArH), 2.48 (3H. s, CH3).
3l: IR (KBr): nmax
3082, 1678, 1600, 1562, 1504, 1345, 1168, 794, 728, 696 cm-1; 1H NMR (CDCl3): d H 9.03 (1H, s, HC=N), 8.02 (2H, d,
J=7.6 Hz, 2,6-SO2ArH), 7.96 (1H, s, 2'-ArH), 7.81 (1H, d, J=7.6 Hz, 6'-ArH), 7.67 (1H, d,
J=7.6 Hz, 4'-ArH), 7.59 (3H, m, 3,4,5-SO2ArH), 7.48 (1H, t, 5'-ArH).
3p: IR (KBr): nmax
3036, 1684, 1622, 1580, 1546, 1320, 1174, 780, 765, 716, 692 cm-1; 1H NMR (CDCl3):
d H 8.82 (1H, d, J=9.2 Hz,
HC=N), 8.00 (2H, d, J=7.2 Hz, 2,6-SO2ArH), 7.66 (1H, d, J=14.8 Hz, PhCH=), 7.58
(5H, m, 2'-6'-ArH), 7.47 (3H, m, 3,5-SO2ArH), 7.00 (1H, dd, J=14.8 Hz and J=9.2
Hz, =CHCHN).
REFERENCES
[1] Prakash, G K S, Mandal, M, Olah, G A. Nucleophilic Synlett, 2001, 77-78.
[2] Davis, F A, Reddy, R T, Weismiller, M C. J. Am. Chem. Soc., 1989, 111: 5964-5965.
[3] Boger, D L, Corbett, W L, Curran, T T, et al. J. Am. Chem. Soc., 1991, 113: 1713-1729.
[4] Boger, D L, Curran, T T. J. Org. Chem., 1990, 55: 5439-5442.
[5] Sisko, J, Weinreb, S M. J. Org. Chem. 1990, 55: 393-395.
[6] Melnick, M J, Freyer, A J, Weinreb, S M. Tetrahedron Lett., 1988, 29: 3891-3894.
[7] Boger, D L, Corbett, W L. J. Org. Chem., 1992, 57: 4777-4780.
[8] Jennings, W B, Lovely, C J. Tetrahedron, 1991, 47: 5561-5568.
[9] Jennings, W B, Lovely, C J. Tetrahedron Lett., 1988, 29: 3725-3728.
[10] Davis, F A, Lamendola, J J, Nadir, U. J. Am. Chem. Soc., 1980, 102: 2000-2005.
[11] Andras, V, Jozsef, D, Rajender, S V. Tetrahedron Lett., 1999, 40: 4951-4954.
[12] Trost, B , Marrs, C J. Org. Chem., 1991, 56: 6468-6470.
[13] Georg, G I, Harriman, G C B, Peterson, S C A. J. Org. Chem., 1995, 60: 7366-7368.
[14] Chemla, F, Hebbe, V, Normant, J F. Synthesis, 2000, 75-77.
[15] Guo, J J, Jin, T S, Zhang, S L, et al. Green Chemistry, 2001, 3: 193-195.
[16] Jin, T S, Ma, Y R, Li, Y, et al. Synth. Commun., 2001, 31: 2051-2054.
[17] Jin, T S, Feng, G l, Yang, M N, et al. Synth. Commun., 2004, 34: 1277-1283.
[18] Jin, T S, Feng, G l, Yang, M N, et al. Chem. Res.(s), 2003, 591-593.
SiO2-OSO3H催化合成芳香磺酰亚胺
(邯郸学院化学系, 邯郸 056005)
摘要 芳香醛与芳香磺酰胺在SiO2-OSO3H固体酸催化剂的催化下合成了一系列芳香磺酰亚胺化合物。 此方法具有反应快、产率高、操作简便等优点。
关键词 芳香磺酰亚胺, 芳香醛, 芳香磺酰胺,固体酸