http://www.chemistrymag.org/cji/2006/083016pc.htm
Mar. 2, 2006 Vol.8 No.3 P.16 Copyright ![]()
http://www.chemistrymag.org/cji/2006/083016pc.htm |
Mar. 2, 2006 Vol.8 No.3 P.16 Copyright |
Synthesis and inhibitory activity studies of 1'-position substituted fidarestat derivatives
Wang Dong1 Pan Jingmiao2
Guo Zengshan1 Zheng Rong1 Zhu Changjin1*
(1School of Chemical Engineering and Environment, Beijing Institute of
Technology, Beijing, 100081 2 School of Life Science and Technology, Beijing
Institute of Technology, Beijing, 100081)
Abstract Fidarestat is known to bind at
the active site of aldose reductase (AR) and to be an excellent inhibitor of the enzyme.
In order to determine the structure-activity relationships of fidarestat and develop more
effective AR inhibitors, a number of compounds with carboxylic acid methyl substituent at
1'-position of fidarestat skeleton have been synthesized, and their ability of AR
inhibition was investigated in the present studies. In the synthetic work, the effect of
bases, which are used as catalysts, to the reaction between the cyclic imides including
polyamide and the methyl bromoacetate was studied. Mono-substituted acetate products were
synthesized with high yields when weak base such as potassium carbonate was used as
catalyst. The introduction of substitutent groups to N-1' position
of the fidarestat skeleton led to almost completely loss of the inhibitory activity. This
experiment has strongly demonstrated the result from crystallographic study of the
AR-fidarestat complex. The studies of inhibitory activity of different hydantoin ARIs
substituted by acetic acid residue can greatly benefit the structure-based drug design of
ARIs.
Keywords aldose reductase inhibitors, fidarestat, carboxylic acid
methyl, synthesis,inhibitory activity
王 栋1 潘经淼2 郭增山1
郑荣1 朱长进1*
(1北京理工大学化工与环境学院,北京,100081 2北京理工大学生命科学与技术学院,北京,100081)
摘要 Fidarestat是一个已知的结合在醛糖还原酶(AR)活性部位的很有效的醛糖还原酶抑制剂(ARIs)。为了研究它的结构-活性关系并发现更强的ARIs,本文合成了几个fidarestat
1’-位羧甲基取代的化合物,并测试了它们的抑制性。在合成过程中,研究了不同的碱做催化剂对含有环状酰亚胺以及多个酰胺键的两种化合物与溴乙酸甲酯反应的影响。用弱碱如碳酸钾高效地合成了单取代乙酸酯产物。所合成以fidarestat为骨架的
1’-位取代物都基本失去了抑制性, 这一实验结果为前人对于AR-fidarestat复合物晶体学研究的结果提供了强有力的证据。所分析的不同海因类抑制剂用乙酸链取代后的化合物的抑制性可以为以结构为基础的ARIs药物设计提供很好的实验基础。
关键词 醛糖还原酶抑制剂 fidarestat 羧甲基 合成 抑制性
众所周知,糖尿病已成为一种严重危害人类健康的疾病。而由糖尿病引起的并发症例如白内障,视网膜病变,神经病变,肾脏病变和动脉粥样硬化也同样严重威胁人的健康。醛糖还原酶抑制剂(ARIs)已被证明是一种很有效的治疗糖尿病并发症的药物[1-3]。过去30年中,至少有14种ARIs被证明非常有效并且已通过II期临床[4]。其中最有效的是在日本已上市的Epalrestat,
和已进入III期临床的Fidarestat 和AS-3201。ARIs及其药物应用研究进展很快,但仍不能满足发展更快的市场对药物的期待与需求[5]。
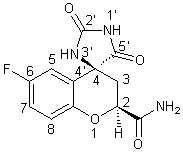
以前的研究表明fidarestat (图1)在体内的抑制效果要远远高于在日本已上市的Epalrestat并且没有观察到副作用[6,
7],这证明它是一个很有潜力的抑制剂。但对于这一非常有潜力的抑制剂,它的结构-活性关系研究甚少,仅仅Mitsuru
Oka等人确定了AR-fidarestat复合物晶体结构,并且分析了二者之间可能的作用[8,
9]。但至今仍没有任何直接的实验证据能证明Mitsuru Oka等人所分析的结果。为了研究fidarestat的结构-活性关系并发现更强的ARIs,我们合成了它的衍生物,通过研究其衍生物的抑制性来检验他们所分析的结果。我们在fidarestat
1’-位用乙酸链取代,设计了四个化合物,所引入的乙酸负离子有可能与AR有化学键形成。因为我们发现很多羧酸类ARIs都含有羧甲基,而这一酸性基团正是提供了酸性质子这一ARIs的必需结构要求
[10]。并且以前的文献报道过类似的用羧酸链取代后衍生物的抑制性(图2),其结果表明对于R’相同,R不同的化合物,R=CH2COOH
时,活性最高[11]。
 |
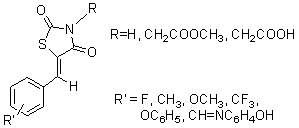 |
图1
Fidarestat 1 |
图2
用不同取代基取代酰亚胺键后形成的醛糖还原酶抑制剂 |
在Fidarestat结构中有3个酰胺键,要达到只对其中一个较为活泼的酰胺进行取代,必须要控制反应条件。本文报道了通过控制碱的种类与用量选择性地取代1’-位的酰胺键,高产率地得到目标产物。这种方法不仅仅对Fidarestat是可行的,还对很多含有相似的螺海因环的ARIs (Sorbinil,Methosorbinil,文献[12] 中的ARIs)都是很有效的。其后又对所合成化合物进行了活性测试与研究。
1. 实验部分
1.1 主要仪器与试剂
核磁:日本JEOL JNM-AL300 型核磁共振波谱仪(TMS为内标,DMSO-d6作溶剂)。质谱
(ESI):Bruker ESQUIRE-3000 型。质谱(FAB): 英国VG ANALYTICAL公司 VG-ZAB-HS型。紫外:瑞利公司UV-1201型紫外分光光度计。
TLC: 60 F254型Merck薄层板。
D,L-甘油醛购于Sigma, NADPH(还原型辅酶II)购于Roche。其余试剂均为市售分析纯,用前未经处理。
我们根据以前的文献[13],以对氟苯甲醚和马来酸酐为原料,合成了Fidarestat
以及化合物2。
1.2 目标产物的合成
目标产物4, 6, 7, 9的合成路线以及化合物表征数据如图3和表4所示。
1.2.1 (2S,4S)-6-氟-2',5'-二氧螺[苯并二氢吡喃-4,4'-咪唑烷]-2-酰胺基-乙醇(3)的合成
把NaH (0.352 g, 8.8 mmol, 60%含量) 溶于80 ml
无水甲醇中,然后向其中逐滴加入乙醇胺(0.49 ml, 8 mmol)。室温搅拌1 h后,加入2
(1.288 g, 4 mmol)。反应液室温搅拌12 h后浓缩到1/10体积,用稀盐酸调酸性到pH=5-6.
产物3作为固体析出,滤出并用水洗涤。再用3×20ml乙酸乙酯萃取母液,无水硫酸钠干燥有机相,减压蒸馏。蒸干后的固体与滤出的固体合并,用乙醇重结晶,得到无色晶体3
(1.09g, 85%)。
1.2.2 (2S,4S)-6-氟-1'-羧甲酯甲基-2
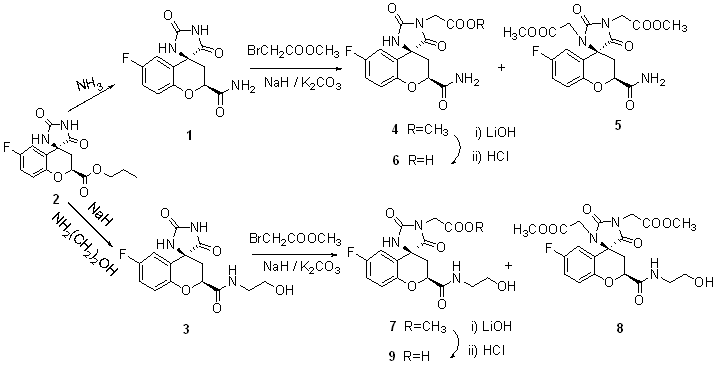
1.2.3 (2S,4S)-6-氟-1'-羧甲基-2',5'-二氧螺[苯并二氢吡喃-4,4'-咪唑烷]-2-酰胺
(6) 的合成
化合物4 (265 mg)溶于15 ml DMF中,向其中加入7.5 ml
1 N LiOH 水溶液,室温搅拌30 min 后停止反应。然后用稀盐酸调酸性到pH=1~2.
接着对水相用3×20ml
乙酸乙酯萃取,无水硫酸钠干燥有机相,减压蒸去有机溶剂。最后柱层析(CHCl3:CH3OH:CH3COOH=100:20:8)对粗产品纯化得240
mg化合物6,产率94%。
1.2.4 (2S,4S)-6-氟-1'-羧甲酯甲基-2',5'-二氧螺[苯并二氢吡喃-4,4'-咪唑烷]-2-酰胺-乙醇
(7) 以及 (2S,4S)-6-氟-1',3'-二羧甲酯甲基-2',5'-二氧螺[苯并二氢吡喃-4,4'-咪唑烷]-2-酰胺-乙醇
(8) 的合成
化合物3、NaH或K2CO3用DMF溶解,向溶液中逐滴加入溴乙酸甲酯,室温搅拌一定时间后(搅拌时间见表2所示)停止反应,用稀盐酸调酸性到pH=5-6。加入10
ml水,3×20ml乙酸乙酯萃取水相,无水硫酸钠干燥有机相。减压蒸干有机相,柱层析(CHCl3:CH3OH
=15:1)对粗产品纯化得到化合物7(Rf = 0.21)和化合物8(Rf
= 0.37)。
1.2.5 (2S,4S)-6-氟-1'-羧甲基-2',5'-二氧螺[苯并二氢吡喃-4,4'-咪唑烷]-2-酰胺-乙醇
(9) 的合成
化合物7(243 mg)溶于15 ml CH3OH中,向其中加入6.2
ml 1 N LiOH 水溶液,室温搅拌30 min
后停止反应。然后用稀盐酸调酸性到pH=1-2。 接着对水相用3×20 ml
乙酸乙酯萃取,无水硫酸钠干燥有机相,减压蒸去有机溶剂。柱层析(CHCl3:CH3OH:CH3COOH
=100:20:8)对粗产品纯化得到化合物9,产率77%。
1.3 酶实验
1.3.1 酶的提取
按照文献报道的方法[14],醛糖还原酶(ALR2, EC
1.1.1.21)从老鼠眼球的晶状体中经部分提纯而得。
从正常杀死的老鼠(Male Wistar mice, Certificate No SCXK
2000-0006, 重量 190-210 g b.w.)眼球中迅速取出晶状体,然后在Glas-Potter匀浆器中,加入3倍(0.4ml/lens)于其体积的冷去离子水(0-4
1 |
溴乙酸甲酯 |
NaH |
K2CO3 |
时间/h |
4 |
5 |
4(equiv.)/ 5(equiv.) |
4产率 |
1 |
2 |
2 |
---- |
0.25 |
0.5 |
0.25 |
2:1 |
50 |
1 |
1.2 |
1 |
---- |
0.25 |
0.53 |
0.20 |
2.65:1 |
53 |
1 |
1.2 |
----- |
1 |
1.5 |
0.88 |
0.047 |
19:1 |
88 |
1 |
1.2 |
----- |
---- |
24 |
0 |
0 |
------ |
0 |
3(equiv.) |
溴乙酸甲酯 (equiv.) |
NaH(equiv.) |
K2CO3(equiv.) |
时间/h |
7(equiv.) |
8(equiv.) |
7(equiv.)/8(equiv.) |
7(equiv.)/回收3(equiv.) |
7产率(%) |
1 |
2 |
2 |
--- |
0.25 |
0.42 |
0.086 |
5:1 |
--- |
42 |
1 |
1.2 |
1 |
---- |
0.25 |
0.44 |
0.080 |
5.5:1 |
--- |
44 |
1 |
1.2 |
---- |
1 |
10 |
0.77 |
0 |
--- |
4.3:1 |
77 |
1 |
1.2 |
---- |
2 |
24 |
0.78 |
0 |
--- |
4.5:1 |
78 |
两种不同的碱所得结果也有相似之处:无论是K2CO3
还是NaH,
单取代产物的产量均明显高于双取代产物。这主要是由于1'-位的胺基是碳二酰亚胺,有两个羰基都会吸引电子,胺基上的氢很活泼,较易离去。而3'-位的胺基为一般的酰胺键,不如1'-位胺基上的氢活泼。推测其反应机理是在反应过程中先大量地生成了1'-位取代的单取代产物,然后这些单取代产物中又有一部分在3'-位取代,生成双取代产物。K2CO3的碱性比NaH弱很多,这样使得3'-位取代更难进行,从而只是生成了1’-位取代的单取代产物。尽管3'-位的胺基氢较为活泼,但仍需要碱催化,如表1所示,在完全没有碱催化时,反应不进行。并且如表2所示,当用类似K2CO3的弱碱时,尽管完全没有副产物8生成,但反应进行很不充分。反应时间延长到24
h 仍未进行完全,所回收的原料占了粗产物将近 20%的比例。因此做这类型反应时,K2CO3碱性正好适中。当所用碱的碱性强于它时,有二取代产物生成;当所用碱的碱性弱于它时,反应很难进行。
2.2 活性研究
我们把从老鼠眼睛提出的AR部分提纯,然后测试了新合成化合物4,6,7,9在体外抑制AR活性的能力(表3),使用fidarestat作为对照化合物。
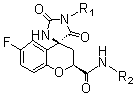
化合物 |
R1 |
R2 |
抑制百分数 (%) |
IC50a |
||||
10-8M |
10-7M |
10-6M |
10-5M |
10-4M |
||||
4 |
CH2COOCH3 |
H |
10 |
14 |
17 |
20 |
27 |
>100 |
6 |
CH2COOH |
H |
0 |
0 |
0 |
8 |
57 |
72 |
7 |
CH2COOCH3 |
CH2CH2OH |
0 |
0 |
4 |
34 |
57 |
49 |
9 |
CH2COOH |
CH2CH2OH |
0 |
0 |
0 |
12 |
50 |
100 |
fidarestat |
H |
H |
38 |
52 |
64 |
70 |
--b |
0.088 |
从所测结果(表3)可以看出,用羧甲酯甲基在fidarestat
1’-位取代后形成的化合物4 (IC50 > 100 mM)以及其相应的羧酸6(IC50
=72 mM)相对于它们的母体化合物fidarestat(IC50=0.088
mM)活性大大降低,几乎没有活性,尽管羧酸6的活性比其相应的酯4好,但它在低浓度时几乎没有抑制,只在10-4才有57%的抑制(如表3所示)。为了更充分地研究N-1’位取代物的抑制性,我们又测试了fidarestat的类似物3的N-1’位取代物的抑制性。从所测结果看出,化合物7与9的失活(其IC50
= 49 mM,100 mM)也很明显。活性结果证明1’-位取代的衍生物抑制性均大大降低。
Mitsuru Oka等人对于AR-fidarestat复合物晶体学研究的结果指出:海因环上1’-位的N原子与AR活性部位His110的N![]() 2有氢键形成[8, 9]。我们认为,在fidarestat海因环的1’-位引入羧酸链时,破坏了原本1’-位N原子与His110的氢键,而AR活性部位体积有限,使得羧酸链指向了AR空腔的入口部位,远离了酶的活性部位。因此,羧酸链取代后的产物由于没有与酶的活性部位的His110,
Trp111 和 Tyr48
残基有相互作用而导致大大失活。我们也可以看到螺海因环对fidarestat的抑制性起了关键性的作用。我们的这一实验结果为前人对于AR-fidarestat复合物晶体学研究的结果[8,9]提供了强有力的证据。
2有氢键形成[8, 9]。我们认为,在fidarestat海因环的1’-位引入羧酸链时,破坏了原本1’-位N原子与His110的氢键,而AR活性部位体积有限,使得羧酸链指向了AR空腔的入口部位,远离了酶的活性部位。因此,羧酸链取代后的产物由于没有与酶的活性部位的His110,
Trp111 和 Tyr48
残基有相互作用而导致大大失活。我们也可以看到螺海因环对fidarestat的抑制性起了关键性的作用。我们的这一实验结果为前人对于AR-fidarestat复合物晶体学研究的结果[8,9]提供了强有力的证据。
但并非所有的羧酸链取代的海因类抑制剂都失活。如前所述,据报道[11],如图2所示在N-1位用乙酸链取代形成的衍生物的活性(R
= CH2COOH > R = H)大大增强。然后他们用计算机模拟,从理论上研究了可能的原因。他们发现所引入的羧基与NADP+的氮正离子有作用,并且同AR活性部位的一个氨基酸Tyr48形成了氢键。因此,使得羧基取代后的化合物活性提高[11]。但从电脑上对接的方法所得出的结论并不能给出他们所设计的化合物其中有一个酯比相应羧酸活性高的原因。
这两类羧酸链取代衍生物活性相对于相应母体化合物活性截然相反,我们推测可能的原因是:当母体化合物体积较小时(如图2所示,仅有一个苯环与非相邻的五元环),所引入的乙酸链容易伸入AR的活性部位。尽管羧酸链的取代会影响这个N原子的氢键,但羧酸负离子能与AR活性部位形成更强的氢键,从而活性增强。当母体化合物体积较大时(如图1所示,fidarestat含有一个大的苯并吡喃环与一个紧邻的五元环)由于AR活性部位体积有限,使得乙酸链羧基指向了AR空腔的入口部位,远离了酶的活性部位。因此,乙酸链取代后的产物由于没有与酶的活性部位有相互作用而导致大大失活。总之,对于母体较小的海因类抑制剂在二酰亚胺位用乙酸链取代后抑制性增强,对于母体较大的海因类抑制剂在二酰亚胺位用乙酸链取代后几乎丧失抑制性。
3. 结论
用含有类似Fidarestat海因螺环化合物做反应原料,要仅得到1’位取代产物时应选用K2CO3做催化剂。在一定范围内碱与反应物的摩尔当量对反应性几乎没有影响,而碱的碱性对反应影响很大。底物:BrCH2COOCH3:
K2CO3 =1: 1.2: 1 是较为合适的反应条件。用羧酸链在fidarestat及其类似物1’-位取代后的衍生物(化合物4,6,7,9)都几乎失活。尽管所合成化合物都几乎失活,但我们的这一实验结果为前人对于AR-fidarestat复合物晶体学研究的结果[8,
9]提供了强有力的证据。所分析的不同海因类抑制剂用乙酸链取代后的化合物的抑制性可以为以结构为基础的ARIs药物设计提供很好的实验基础。
化合物 |
d H |
m/z |
3 |
2.04 (t, 1 H, J = 12, CCH2CH), 2.44 (dd, 1 H, J = 12 and 3, CCH2CH), 3.19 (dd, 2 H, J = 12 and 6, CH2CH2OH), 3.43 (dd, 2 H, J = 12 and 6, CH2CH2OH), 5.08 (dd, 1 H, J = 12 and 3, CH2CHO), 6.96-7.19 (m, 3 H, Ph), 8.22-8.26 (m, 1 H, CONHCH2), 8.41 (s, 1 H, CNHCO), 11.03 (s, 1 H, CONHCO) |
322.0 ESI |
4 |
2.10 (t, 1 H, J = 12, CCH2CH), 2.47 (m, 1 H, CCH2CH), 3.69 (s, 3 H, COOCH3), 4.25 (d, 2 H, J = 2, NCH2), 4.97 (dd, 1 H, J = 12 and 3, CH2CHO), 6.96-7.22 (m, 3 H, Ph), 7.57 (s, 1 H, CONH2), 7.79 (s, 1 H, CONH2), 8.94 (s, 1 H, CNHCO) |
352.5 ([M+H]+) FAB |
5 |
2.37 (m, 2 H, CCH2CH), 3.62 (s, 3 H, COOCH3), 3.69 (s, 3 H, COOCH3), 3.81 (d, 1 H, J = 18, NCH2), 4.23 (d, 1 H, J = 18, NCH2), 4.34 (s, 2 H, NCH2), 5.01 (dd, 1 H, J = 12 and 3, CH2CHO), 6.96-7.24 (m, 3 H, Ph), 7.61 (s, 1 H, CONH2), 7.71 (s, 1 H, CONH2) |
424.1 ([M+H]+) FAB |
6 |
2.10 (t, 1 H, J = 12, CCH2CH), 2.47 (m, 1 H, CCH2CH), 4.14 (d, 2 H, J = 2, NCH2), 4.98 (dd, 1 H, J = 12 and 3, CH2CHO), 6.96-7.22 (m, 3 H, Ph), 7.56 (s, 1 H, CONH2), 7.77 (s, 1 H, CONH2), 8.68(s, 1 H, CNHCO) |
337.5 ([M+H]+) FAB |
7 |
2.14 (t, 1 H, J = 12, CCH2CH), 2.46 (dd, 1 H, J = 12 and 3, CCH2CH), 3.21 (dd, 2 H, J = 12 and 6, CH2CH2OH), 3.44 (dd, 2 H, J = 12 and 6, CH2CH2OH), 3.69 (s, 3 H, COOCH3), 4.25 (d, 2 H, J = 2, NCH2), 5.03 (dd, 1 H, J = 12 and 3, CH2CHO), 6.96-7.23 (m, 3 H, Ph), 8.25-8.31 (m, 1 H, CONHCH2), 8.94 (s, 1 H, CNHCO) |
396.4 ([M+H]+) FAB |
8 |
2.37 (m, 2H, CCH2CH), 3.21 (dd, 2 H, J = 12 and 6, CH2CH2OH), 3.44 (dd, 2 H, J = 12 and 6, CH2CH2OH), 3.62 (s, 3 H, COOCH3), 3.69 (s, 3 H, COOCH3), 3.81 (d, 1H, J = 18, NCH2), 4.16 (d, 1 H, J = 18, NCH2), 4.33 (s, 2 H, NCH2), 5.08 (dd, 1 H, J = 12 and 3, CH2CHO), 6.97-7.24 (m, 3 H, Ph), 8.17-8.21 (m, 1 H, CONHCH2) |
468.1 ([M+H]+) FAB |
9 |
1.98 (t, 1 H, J = 12, CCH2CH), 2.19 (dd, 1 H, J = 12 and 3, CCH2CH), 3.28 (dd, 2 H, J = 12 and 6, CH2CH2OH), 3.51 (dd, 2 H, J = 12 and 6, CH2CH2OH), 4.08 (d, 2 H, J = 2, NCH2), 5.12 (dd, 1 H, J = 12 and 3, CH2CHO), 6.97-7.24 (m, 3 H, Ph), 8.29-8.35 (m, 1 H, CONHCH2), 8.87 (s, 1 H, CNHCO) |
382.2 ([M+H]+) FAB |
Synthesis and inhibitory activity studies of 1'-position substituted fidarestat derivatives
Wang Dong1 Pan Jingmiao2
Guo Zengshan1 Zheng Rong1 Zhu Changjin1*
(1School of Chemical Engineering
and Environment, Beijing Institute of Technology, Beijing, 100081 2 School of
Life Science and Technology, Beijing Institute of Technology, Beijing, 100081)
According to previous literatures, when
p-fluoroanisole and maleic acid were used as starting material, compounds 1 and 2
could be synthesized. Then compound 2 was aminolyzed by ethanoamine in the
presence of sodium hydride to provide compound 3. Mono-substituted compound 4
were prepared by the reaction between N-unsubstituted compound 1 and methyl
bromoacetate, using sodium hydride or potassium carbonate as base. However, tremendous
different yields were observed by using the two different bases. When the former base was
used, the bis-substituted byproduct 5 was formed in a higher percentage. In
contrast to this, mono-substituted compound 4 was obtained in a much higher yield
in the presence of potassium carbonate. The similar result was also seen in the synthesis
of compound 7. The acids 6 and 9 could be prepared by hydrolyzing the
corresponding esters 4 and 7, respectively. The loss of inhibition activity
of compounds 4, 6, 7 and 9 compared to the reference compound,
fidarestat, has strongly demonstrated the
result from crystallographic study of the AR-fidarestat complex.