http://www.chemistrymag.org/cji/2006/085032pe.htm |
May 1, 2006 Vol.8 No.5 P.32 Copyright |
Sun
Hanwen, He Pan, Lv Yunkai, Zhao Xiaoli, Liang Shuxuan
(College of Chemistry and Environmental Sciences, Hebei University, Key Laboratory of
Analytical Science and Technology of Hebei Province, Baoding 071002, China)
Abstract A new method was described
for simultaneous determination of ciprofloxacin (CIP), ofloxacin (OFL) and norfloxacin
(NOR) by capillary electrophoresis with a diode array detector at 280 nm. The separation
conditions were investigated and optimized. A new medium, 30 mM sodium tetraborate
buffer solution and 22 mM sodium dodecyl sulphate containing 5% (v/v) methanol (pH 8.7),
was developed for the complete separation of the three quinolones. The excipients do not
affect to separation and analysis. Calibration curves between peak area and concentration
in the range 10-1000 mg/L for each compound have good linearity with a correlation
coefficient (r) of greater than 0.999, and the detection limit(s/n) was 7.90 mg/L for
CIP,7.92 mg/L for OFL and 7.56 mg/L for NOR. This
method has been successfully applied for simultaneous determination of CIP, OFL and NOR in
the pharmaceutical preparations with rapid, accurate, and simple characteristics.
Keywords Capillary electrophoresis; Ciprofloxacin; Ofloxacin; Norfloxacin;Pharmaceutical analysis
1 INTRODUCTION
Fluoroquinolones are chemotherapeutic agents that are widely used in human and
veterinary medicine because of their safety with good tolerance and broad antibacterial
spectrum, which exhibit bactericidal activity primarily by inhibiting bacterial DNA
gyrase. Ciprofloxacin (CIP), ofloxacin (OFL) and norfloxacin (NOR) are three most
important and widely used fluoroquinolones, and have a great potency against many common
bacterial pathogens and high activity against Gram-negative and Gram-positive bacteria in
vivo and in vitro.
As far as we know, many analytical methods concerning fluoroquinolones
have been published. Most of them are based on high-performance liquid chromatography
(HPLC) [1-5]. However, capillary electrophoresis (CE) has become a useful technique for
pharmaceutical analysis because of its high separation efficiency, high resolution, high
speed and small sample volume requirements. Some methods have been developed for the
determinations of CIP, OFL and NOR by CE, respectively [6-8]. Simultaneous determination
is rapidly growing in fluoroquinolone pharmaceutical analysis [9-15]. Josephine et al.
described a capillary zone electrophoresis–electrospray
ionization tandem mass spectrometry method for the analysis of fluoroquinolone
antibiotics. Nine fluoroquinolones have been separated and detected in a single run by
this technique [9]. However, the detector cannot be found in common lab. Simultaneous
determination of 14 quinolone antibacterials was achieved by CE using overlapping
resolution mapping scheme, but the separation system is complicated [10]. Fierens et al.
used a phosphate buffer (pH 7.0, 125 mM) to study the analysis of 10 quinolones, but
norfloxacin and ciprofloxacin cannot be separated in this way [11]. Obviously, a more rapid, less cost and easier operation method is also
required.
The aim of this study was to elaborate and validate a simple and
efficient DAD-CE methods for the separation and simultaneous determination of CIP, OFL and
NOR in pharmaceutical preparations.
2 EXPEREMENTAL
2.1 Apparatus and conditions
All the experiments were performed by an Agilent 3D CE system with air-cooling and a diode
array detector (Agilent Technologies, Waldbronn, Germany). A 48.5 cm (40.0 cm to the
detector) × 50 mm i.d.
uncoated fused-silica capillary (Agilent) was utilized. Other conditions are: capillary
temperature at 20 ℃,applied voltage at 15 kV and
UV detection at 280 nm. The hydrodynamic injection was at a pressure of 50 mbar for 4 s.
The capillary was conditioned at the beginning of each day with 0.1 M NaOH for 5 min,
followed by water for 5 min and running buffer for 10 min. Between consecutive analyses,
the capillary was flushed with water for 1 min and the running buffer for 3 min to
guarantee good reproducibility.
2.2 Chemicals and reagents
All the chemicals used were of analytical reagents: Methanol (MeOH), boric acid, sodium
tetraborate decahydrate, sodium dodecyl sulphate (SDS), sodium hydroxide and hydrochloric
acid were obtained from Beijing Chemical Factory (Beijing, China); Ciprofloxacin,
ofloxacin and norfloxacin were obtained from the National Institute for the Control of
Pharmaceutical and Biological Products (Beijing, China). The structures of the studied
compounds are shown in Fig. 1. Doubly deionized water (DDW) was used throughout.
|
|
|
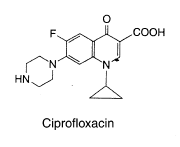
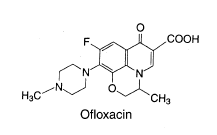
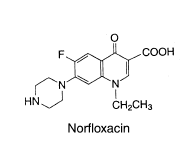
Fig. 1 The chemical structures of ciprofloxacin, ofloxacin and norfloxacin
2.3. Preparation of buffer and
standard solutions
Three stock solutions (2000 mg/L) of CIP, OFL and NOR were prepared by dissolving 20 mg
standards in 0.1 M HCl (10 ml) and stored under refrigeration. Reference solutions for
quantitative analysis of CIP, OFL and NOR were prepared from the commercially available
drugs by mixing the powder with the running buffer.
The electrophoretic solution was 30 mM sodium tetraborate and 22 mM SDS
containing 5% (v/v) methanol at pH 8.7. The buffers with different concentrations were
adjusted to the desired pH-values with 1 M NaOH and 1 M HCl. The suspensions were filtered
through a membrane (0.22
3. RESULTS AND DISCUSSION
3.1. Optimization of capillary electrophoresis separation
The aim of our experiments was to develop a simple and rapid method for the separation of
CIP, OFL and NOR by CE. The pKa values of the three compounds are shown in Table 1[16].
| Analyte | pKa (-COOH) | pKa (≡NH+) |
| CIP | 6.27 |
8.87 |
OFL |
5.97 |
7.65 |
| NOR | 6.26 |
8.85 |
The given pKa corresponds to acidity of the carboxyl group and the
nitrogen located at the distant end of the piperazine. The best separation based on this
parameter is expected to be found at a pH (around 8.87), where all the compounds would be
analyzed as anions. If analysis was carried out in 30 mM sodium tetraborate at different
pH values, OFL, CIP and NOR could not be separated completely, as shown in Fig.2.
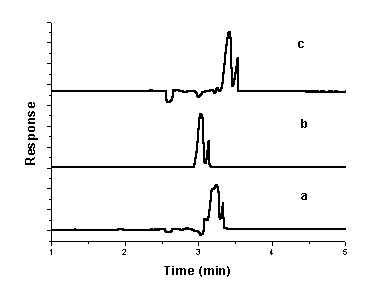
Fig. 2 Electropherogram of a mixture of CIP,
OFL and NOR when using 30 mM sodium tetraborate at different pH values (a) pH 8.4;(b) pH 8.7;(c) pH 9.0;
injection, 50 mbar for 4 s; separation voltage, 15 kV; capillary temperature, 20oC ; DAD
detection at 280 nm.
The
separation of CIP and NOR would be improved using the micellar electrokinetic
chromatography (MEKC), which is based on the relative distribution of the analytes between
the aqueous phase and the micellar phase added to the separation buffer (hydrophobicities
of the analytes). SDS is the most routinely used anionic surfactant for the analysis of
fluoroquinolones with MEKC [15-16]. At a concentration over the critical micellar
concentration (CMC), spontaneous aggregation of the surfactant molecules is caused by the
increase of hydrophobic interactions. The working range was set at 18–28 mM of SDS (Fig. 3), and the critical concentration of
22 mM SDS was chosen, which gave the better results, as shown in Fig. 3(b). The
migration order of these compounds is thus governed by a combined effect of their
hydrophobicities (incorporation into the hydrophobic sites of the micelles) and their
positive charge (ionic interactions between the negatively charged micelle surface and the
cationic part of the analytes).
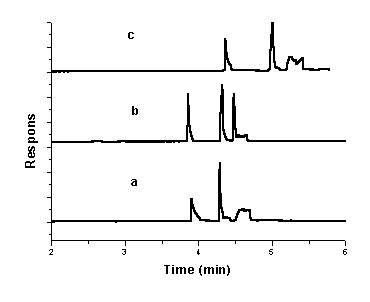
Fig.3 Electropherogram of a mixture
of CIP, OFL and NOR with (a) 18 mM;(b) 22 mM;(c) 28 mM SDS in 30 mM sodium tetraborate (pH 8.7); injection, 50
mbar for 4 s; separation voltage, 15 kV; capillary temperature, 20oC ; DAD detection at
280 nm.
From Fig. 3, it can be seen that CIP, OFL and NOR were
completely separated. But the shape of the third peak was still bad.
Organic solvents added to the running buffer can improve the shape of the third peak,
which is due to the addition of solvents with a high dielectric constant and lipophilic
character (lipophilic character increasing the solubility of the monomeric surfactant and
the CMC value). Methanol is a typical protic solvent with little lipophilic character,
does not appear to disturb the micellar system much, since the increase of the CMC is
small even for volume percentages as high as 35% [17]. When methanol was added to the
running buffer, the migration time of three peaks was increased (Fig. 4).
The best results were obtained when 5% methanol was
added. Three peaks were separated completely and the peaks were very sharp (Fig. 4.b).
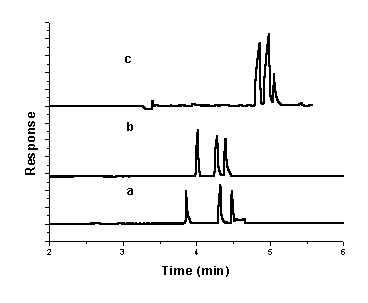
Fig.4 Electropherogram of a mixture
of CIP, OFL and NOR with (a) 0;(b) 5%;(c) 10% MeOH in 30 mM sodium tetraborate and 22 mM SDS (pH
8.7); injection, 50 mbar for 4 s; separation voltage, 15 kV; capillary temperature, 20oC;
DAD detection at 280 nm..
3.2. Quantitative
determination in pharmaceutical preparations
The method was applied for the quantitative determination of CIP, OFL and NOR in the
mixtures of their tablets and capsules. The electropherogram of mixture of CIP, OFL and
NOR in tablets and capsules were shown in Fig. 5.b.
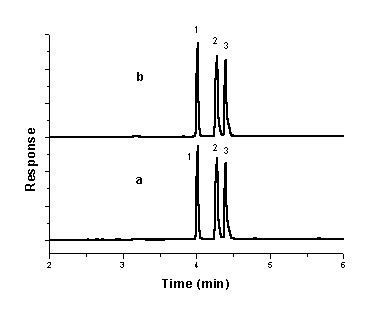
Fig.5 Electropherogram of mixture of
CIP, OFL and NOR (a) in standard solution; (b) in tablets and capsules. A buffer
consisting of 30 mM sodium tetraborate and 22 mM sodium dodecyl sulphate containing 5%
(v/v) methanol (pH 8.7); injection, 50 mbar for 4 s; separation voltage, 15 kV; capillary
temperature, 20oC ; DAD detection at 280 nm. 1. OFL;2.
.NOR;3.CIP
It can be seen that the excipients (maize starch, lactose, magnesium stearate, etc.) in the pharmaceutical preparations do not have adverse effect on the determination of CIP, OFL and NOR.
3.3 Validation of the method
In most HPLC methods, large amount of organic solvent have to be used as mobile phase and
long analytical time is usually a boring problem to be resolved. Comparing with HPLC, the
proposed CE method is simple, fast, and much practical for the determination of CIP, OFL
and NOR with 30 mM sodium tetraborate buffer containing 22 mM SDS solution 5% (v/v)
methanol (pH 8.7) at 20 ℃ and 15 kV. The whole
analysis was achieved within 5 minutes.
The linearity for CIP, OFL and NOR analysis was evaluated with
concentrations of calibration standards against measured peak areas under the optimal
conditions. The equations of calibration curve and detection limits (S / N = 3) were
listed in Table 2.
Table 2 Linearity and detection limits of CIP, OFL and NOR
Analyte |
Linear
range |
Regression equations |
Correlation coefficient |
Detection
limit |
CIP |
10-1000 |
Y=0.6581X-13.6193 |
0.9996 |
7.90 |
OFL |
20-800 |
Y=0.4286X-15.6584 |
0.9993 |
7.92 |
NOR |
20-1000 |
Y=0.4553X-37.8999 |
0.9994 |
7.56 |
It can be seen that the
linearity was satisfactory with a correlation coefficient (r) greater than 0.999,
and the detection limits for three compounds were 7.56-7.92 mg/L.
The mean value of the contents and relative standard deviation (RSD)
were determined under the same operating conditions by determining 10 replicate samples on
the same day. The result is shown in Table 3, The RSD is less than 2 %. The recovery for
CIP, OFL and NOR are shown in Table 4. The recoveries are in the range of 95.3 -104.1%.
Table 3 Precision for determining 10 replicate samples
Pharmaceutical preparations |
Analyte |
Theoretical amount |
Amount found |
RSD (%) |
Tablet |
CIP |
250 |
258.8 |
1.56 |
Tablet |
OFL |
100 |
104.7 |
1.62 |
Capsules |
NOR |
100 |
103.4 |
1.13 |
Table 4 The recoveries of CIP, OFL and NOR
| Analyte | Content in sample | Added (mg/l) | Found (mg/l) | Recovery (%) |
| CIP | 165.6 | 100.0 | 269.7 | 104.1 |
| CIP | 165.6 | 400.0 | 566.6 | 100.3 |
| OFL | 183.3 | 100.0 | 279.7 | 96.4 |
| CIP | 183.3 | 400.0 | 598.8 | 103.9 |
| NOR | 210.1 | 100.0 | 310.4 | 100.3 |
| NOR | 210.1 | 400.0 | 591.2 | 95.3 |
4 CONCLUSION
The results suggest that 30 mM of borate buffer and 22 mM SDS containing
5% (v/v) methanol at pH 8.7 is a very efficient running buffer for separating CIP, OFL and
NOR. The study demonstrates that capillary electrophoresis can be successfully applied for
the qualitative and quantitative analysis of CIP, OFL and NOR in pharmaceutical
formulations.
ACKNOWLEGEMENT
This work was supported by the Specialized Research Funds of China Education Ministry (No.20050075003)REFERENCES
[1] Carlucci G. J. Chromatogr. A,1998, 812, 343–367.
[2] Barbosa J, Berges R, Sanz-Nebot V.J. Chromatogr. A, 1998, 823, 411–422.
[3] Pauline M L, Norman M C, Roger W S. J. Pharm. Biomed. Anal.,1996, 14, 641–654.
[4] Cordoba-Borrego M, Cordoba-Diaz M, Cordoba-Diaz D. J. Pharm. Biomed. Anal., 1999, 18,
919–926.
[5] Samanidou V F, Demetriou C E, Papadoyannis I N. Anal. Bioanal. Chem.,2003, 375, 623-629.
[6] Katarzyna M, Genowefa P, Stefan T. J. Chromatogr. A,2004, 1051, 267–272.
[7] Bannefeld K H, Stass H, Blaschke G. J. Chromatogr. B,1997, 692, 453–459.
[8] Barrón D, Jiménez-Lozano E, Cano J et al., J. Chromatogr. B, 2001759, 73–79.
[9] Josephine M, Guy B, Adela R R.J. Chromatogr. A,2003,
990, 259–269.
[10] Sun SW, Chen L Y. J. Chromatogr. A,1997, 766,
215–224.
[11] Fierens C, Hillaert S, Van den Bossche W. J. Pharm. Biomed. Anal., 2000, 22, 763–772.
[12] Barrón D, Jiménez-Lozano E, Bailac S et al., Anal. Chim. Acta, 2003, 477, 21–27.
[13] Hernández M, Borrull F, Calull M. J. Chromatogr. B 2000, 742, 255–265.
[14] Margarita M, Aguilar C, Borrull F et al., Chromatogr.
B, 2002, 772, 163–172.
[15] Dolores B, Eulalia J L, José B. Anal. Chim. Acta, 2000, 415, 83–93.
[16] Schmitt-Kopplin P h, Burhenne J, Freitag D et al., J. Chromatogr. A,1999, 837,
253-265.
[17] Jacquier J C, Desbene P L. J. Chromatogra. A,1996,
743, 307-314.
孙汉文* 何攀 吕运开 赵晓丽 梁淑轩
(河北大学化学与环境科学学院,河北省分析科学与技术重点实验室,保定 071002)
摘要 提出了一种同时测定环丙沙星、氧氟沙星和诺氟沙星的毛细管电泳分析新方法。以四硼酸钠缓冲溶液(30 mM)-十二烷基硫酸钠(22 mM)- 甲醇(5%,v/v)作为分离介质可使这三种喹诺酮药物得到有效的分离。赋形物不干扰药物的分离与测定。峰面积─药物浓度(10-1000 mg/L)校正曲线具有良好的线性。环丙沙星、氧氟沙星和诺氟沙星的检出限(s/n =3)分别为7.90、7.92、7.56 mg/L。提出的方法具有快速、准确、简易的特点,已成功地应用于药物制剂中的环丙沙星、氧氟沙星和诺氟沙星的测定。
关键词 毛细管电泳;环丙沙星;氧氟沙星;诺氟沙星;药物分析