http://www.chemistrymag.org/cji/2009/113011pe.htm
Mar.1,
2009 Vol.11 No.3 P.11 Copyright ![]()
http://www.chemistrymag.org/cji/2009/113011pe.htm |
Mar.1,
2009 Vol.11 No.3 P.11 Copyright |
Li Lijuna, Li Cuizhea, Zhang Dahaib,
Zang Jiechaoa
(a, College of Chemistry and Environment Science, Hebei University, Baoding 071002; b,
department of chemistry, Handan college, Handan 056005)
Received Nov. 28, 2008; Supported by the fund from the Natural Science Foundation of Hebei Province (No. B2007000152) and Plan Item of Hebei Province Science-Technology Department (05215104)
Abstract A novel 3,3'-BINOL derivative:
3,3'-diethoxycarbonyl-2,2'- diethoxy-1,1'- binaphthyl(2) was synthesized and its structure
was characterized by IR, 1H NMR, 13C NMR and X-ray diffraction.
Keyword 3, 3'-diethoxycarbonyl-2, 2'- diethoxy-1, 1'- binaphthyl, synthesis,
characterization
1 INTRODUCTION
Optically active BINOL and its derivatives are the most prominent C2-symmetric,
axial-chiral ligands��they are
well known to apply in asymmetric catalysis and chiral recognition[1-4]
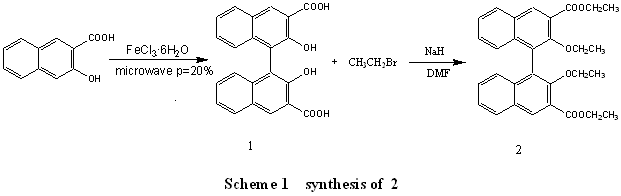
2. EXPERIMENT
2.1 Materials and general methods
Commercial reagents were used without further purification unless otherwise noted. The
melting points were determined using a digital melting point apparatus. IR spectrawere
recorded on a Nicolet AVATAR-37ODTGS Spectrophotometer in the region 4000-400 cm-1 KBr discs. 1H NMR and 13C
NMR spectra were recorded on a Bruker Avance-400 superconduct nuclear magnetic resonance
instrument, in which chemical shift values are reported in ppm (d) relative to TMS as
internal standard. X-ray date was recorded on Rigaku Saturn X ray diffractometer at room
temperature.
2.2 Syntheses of 1
The BINOL-3, 3'-dicarboxylic acid was synthsized
according to reported mothed [9]. Iron(
Table 1. Crystal data and structure refinement for 2
Empirical formula |
C30H30O6 |
q range for data collection | 2.71 to 27.63�� |
Formula weight |
486.54 |
Limiting indices |
-14 < h < 12 |
Temperature (K) |
293(2) |
�� | -20 < k < 20 |
Wavelength(Å) |
0.71073 |
�� | -20 < l < 20 |
Crystal system |
Monoclinic |
Reflections collected |
19223 |
Space group |
P21 /n |
Independent reflections |
5903 (Rint = 0.0397) |
a( Å) |
11.138(18) |
Completeness to q |
97.8 % |
b( Å) |
15.60(3) |
Goodness-of-fit on F2 |
1.061 |
c( Å) |
15.80(3) |
Data / restraints / parameters |
5903 / 0 / 330 |
Volume( Å3) |
2596(7) |
R1(R1 all data) |
0.0915 |
Z |
4 |
wR2 |
0.1980 |
Dcalcd (Mg/m3) |
1.254 |
Largest diff. peak and |
0.267 and -0.241 |
m (mm-1) |
0.086 |
||
F(000) |
1032 |
Table2 Selected bond and distance (
Å) and angles (��) of 2C(1)-C(11) |
1.496(4) |
C(21)-O(6) |
1.204(3) |
C(22)-O(1) |
1.189(3) |
O(4)-O(6) |
2.865 |
O(2)-O(3) |
2.730 |
�� | |
| �� | �� | �� | �� |
C(2)-C(1)-C(11) |
120.62(19) |
C(10)-C(1)-C(11) |
118.56(16) |
C(20)-C(11)-C(1) |
119.03(06) |
C(12)-C(11)-C(1) |
120.16(17) |
C(12)-O(4)-C(27) |
116.34(17) |
C(2)-O(3)-C(25) |
115.63(17) |
O(5)-C(11)-O(6) |
2.246(4) |
O(1)-C(22)-O(2) |
121.6(2) |
2 mL was added to the tube; compounds2
(9.7mg, 0.02mmol) were dissolved in 2ml DMF, was then slowly added along the sides of the
tube via a pipette so that the solution was not disturbed, forming the upper layer. The
tube was then covered and the solvents were allowed to slowly diffuse at room temperature.
After 7 days, colorless prism crystal was found.
The resultant crystals were separated from the solution by decanting.
Suitable X-ray quality crystals were recovered and mounted using epoxy glue. Data were
collected on a Bruker SMART APEX CCD area detector diffractometer. SAINT (Bruker, 1998),
SHELXTL (Bruker, 1998) and SHELXS 97 [11-12] were used for cell refinement,
data reduction and structure solving and refinement of structure. Molecular graphics and
publication materials were prepared using SHELXTL [13]. Details of the crystal
data are listed in Table 1.Selected bond lengths and angles are listed in Table
2.
3 RESULTS AND DISCUSSION
3.1 Syntheses and spectral analyses
Firstly, we synthesized the compound 1 according to reported mothed [9], when setting the
power of microwave oven with 20%, the mole ratio n(Iron(
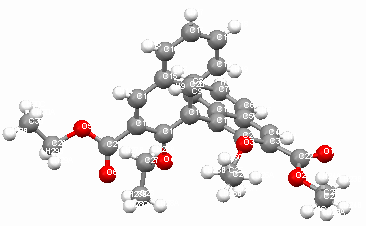
In the crystal cell of 2, every two molecules form a small cuboid channel, four molecules form a bigger cuboid channel, and there are two naphthyl rings are parallel in two adjacent molecules (see Firgure 2 and
Firgure 3).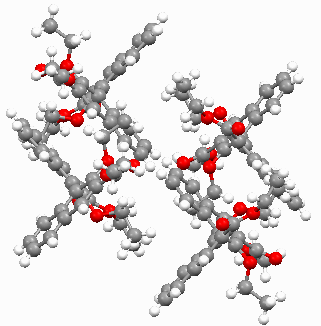
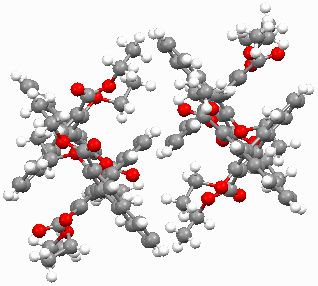
Figure 3 crystal cell of 2 along b axis
REFERENCES
[1] Maki M, Takeshi N. Tetrahedron Lett., 38 (1997): 6233.
[2] Pu L. Chem. Rev., 98 (1998): 2405.
[3] Junji I, Hiroshi F, Tetsuji H. Chem. Rev., 102 (2002): 2211.
[4] Helen C A. Chem. Rev. 102 (2002): 1807.
[5] Haruro I, Yasuhiro Y, Haruka S, etal. J. Am. Chem. Soc., 122 (2000):
5403.
[6] Ana M, Karl H D. Tetrahedron: Asymmetry, 16 (2005): 3256.
[7] Yu C, Shahla Y, Andrei K, etal. Chem. Rev., 103 (2003):3155.
[8 Pavel K, Sÿt