http://www.chemistrymag.org/cji/2009/115026pe.htm |
May.1,
2009 Vol.11 No.5 P.26 Copyright |
Preparation and evaluation of molecularly imprinted monolithic column for liquid chromatographic determination of enrofloxacin in eggs
Lu Yunkai, Liu Yue, Bian Chao, Lu Guodong,
Qin Xinying
(College of Chemistry and Environmental
Science, Hebei University, Key Laboratory of Analytical Science and Technology of Hebei
Province, Baoding 071002, China)
Abstract A molecularly
imprinted HPLC monolithic column for enrofloxacin was prepared by an in situ
thermal-initiated copolymerization technique for enrichment and rapid separation of a
homologous series of fluoroquinolones. The morphological characteristics of the monolithic
molecularly imprinted polymer (monolithic MIP) were investigated by scanning electron
microscope, which showed that both mesopores and macropores were formed in the monolith,
and the monolith allowed the mobile phase to flow through the column with low
backpressure. Furthermore, some chromatographic conditions such as the influence of mobile
phase composition and flow rate on the retention times were investigated. Hydrogen bonding
interaction and hydrophobic interaction played important roles in the retention and
separation. The binding capacity was evaluated by static adsorption and Scatchard
analysis, which showed that the dissociation constant (KD ) and the maximum
binding capacity (Qmax ) were 0.3275 mmol L-1, and 75.88 mmol g-1 for high
affinity binding site, and 8.264 mmol L-1 and 521.5 mmol g-1
for lower affinity binding site, respectively. The present monolithic MIP was successfully
applied to the quantitative determination of ciprofloxacin and enrofloxacin in eggs.
Keywords Molecularly imprinted polymer, Monolithic column,
Enrofloxacin, Food
Molecular imprinting technique (MIT) is increasingly developing for preparing polymers with desired and predetermined selectivity, and provides specific binding sites or catalytic sites in molecularly imprinted polymers (MIPs)[1]. The special binding sites are formed by the self-assembly of a template molecule with specific functional groups in the monomer, followed by a cross-linking polymerization. After removal of the template molecule, the resultant cavities that complement the template in size, shape, and matrix of functional groups from the functional monomer are allowed to rebind the template molecule selectively[2]. The MIP possess several advantages over their biological counterparts including low-cost, simple and convenient preparation, storage stability, repeated operations without loss of activity, high mechanical strength, durability to heat and pressure, and applicability in harsh chemical media [2-4]. Due to such outstanding advantages, they are increasingly being used as selective supports in liquid chromatography[5-9], capillary electophoresis[10], and solid-phase extraction [11-14], and as catalysts bionic sensors [15, 16] and artificial antibodies[17], and membrane separation[18].
Until now, much effort was put on the improvement of the traditional bulk MIP technology by adjusting functional monomers, cross-linking monomers and the shape of the produced particles. An important criterion is the homogeneity of the particles used as column packing material for HPLC. The traditional bulk MIP technology preparing MIPs gives irregular particles with broad size distribution. The traditional bulk MIP technology is to grind the resulting polymer block into particles, and sieve the particles into the desired size ranges. Such ground and sieved particles would be packed into conventional HPLC columns. The particles produced in this tedious and time-consuming process are irregular in size and shape, resulting in significant loss of their chromatographic performance. To overcome these disadvantages, a variety of methods have been developed to make the spherical MIPs, such as swelling method [19,20], suspension polymerization [21], precipitation polymerization [22,23] and surface imprinting on the spherical polymer or silica [24,25], etc.
In 1993, Jun Matsui firstly reported that Monolithic MIP prepared by in situ synthesis technique [26]. An advantage of the technique is its easy handling, a single-step procedure enables us to prepare a column packed with molecular-imprinted polymers without any tedious steps because the polymerization is carried out in the column. Another advantage is its high reproducibility and rapid mass transport. Furthermore, the preparation of this type of MIP is more cost-efficient, because it requires much smaller amount of template molecules. However, the prepared monolithic MIP often suffers from high backpressures and low efficiency, which results in their poor application and practical separation.
The MIP monolith as stationary phase has been successfully used in high performance liquid chromatography to separate enantiomers and analogs, such as the D, L-amino acid derivatives[9, 27], (+)-nilvadipine enantiomers [28], and nicotine [29]. Moreover, this in situ synthesis technique has been applied to prepare MIP stationary phase of capillary electrochromatography for the separation of racemic mixture [30,31]. These studies exhibited that the monolithic MIP possesses high porosity, good permeability and large specific surface area. However, the most of monolithic MIP works were focused on preparation and recognition mechanisms, and applications to real samples are very rarely.
Quinolones are antibiotics agents widely used in the treatment of infectious diseases in both human and animal. It is well known that residues of such antibiotics may persist in edible animal tissues, which makes quinolones potentially hazardous to human health. Thus, it is important to develop a simple and scientific analytical method for determining these drugs present in biomatrices at low levels. Most of the analytical methods established thus far for quinolones are based on liquid chromatography (LC) [32-34].The development of the molecular imprinting technique about quinolone medicament have some study and put up huge potential and advantage [35-38].
The purpose of our work was that the monolithic MIP columns were designed and prepared for preconcentration and rapid separation of a homologous series of fluoroquinolones by in situ thermal-initiated copolymerization technique using enrofloxacin as the template, methacrylic acid (MAA) as the functional monomer and ethylene glycol dimethacrylate (EDMA) as crosslinker. Chromatographic conditions, such as separation mechanism, binding capacity and morphological characteristics of the monolithic column were also investigated and discussed. The present monolithic column was successfully applied for the quantitative determination of ciprofloxacin, and enrofloxacin in eggs with satisfactory results. 2. EXPERIMENTAL
2.1 Chemicals
Methacrylic acid (MAA), Methyl methacrylate (MMA), Ethylene glycol dimethacrylate (EDMA), Acrylamide (AM) and 4-Vinylpyridine (4-VPY) were purchased from Sigma-Aldrich (Shanghai), Shanghai experiment reagent Co., Ltd. (Shanghai, China), Trading Co., Ltd.(Shanghai, China) respectively and distilled under vacuum to remove the stabilizers prior to used. Enrofloxacin (ENR) and ciprofloxacin (CIP) was obtained from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China), their structures are shown in Fig. 1. 2, 2-Azobisisobutyronitrile (AIBN) was purchased from Beijing Chemical Reagent Company (Beijing, China) and recrystallized from methanol before use. All the other chemicals were of the analytical or the HPLC grade and used without further disposal.
Stock solutions (0.5
mg/mL): Transfer 5.0 mg ENR to 50 mL brown volumetric flask, dissolve each in MeOH, and
then dilute to volume with MeOH. Transfer 5.0 mg CIP to 50 mL brown volumetric flask,
dissolve each in 0.01M NaOH, and then dilute to volume with 0.01 M NaOH. Store
refrigerated. Prepare fresh every 6 months.
Table 1 The optimization of the preparation conditions for ENR-MIP monolithic
column
Polymer |
Amount (mmol) |
k'ENR |
k' CIP |
a |
Rs |
IF |
|||
Template |
MAA |
MMA |
EDMA |
�� | �� | �� | �� | �� | |
P1 |
0.5 |
1.5 |
0 |
10 |
4.34 |
2.14 |
2.03 |
0.43 |
2.04 |
P2 |
0 |
1.5 |
0 |
10 |
2.13 |
2.13 |
1 |
�� |
�� |
P3 |
0.5 |
2.0 |
0 |
10 |
4.83 |
2.2 |
2.2 |
0.55 |
2.40 |
P4 |
0 |
2.0 |
0 |
10 |
2.01 |
2.01 |
1 |
�� |
�� |
P5 |
0.5 |
3.0 |
0 |
10 |
4.12 |
2.15 |
1.92 |
0.38 |
1.94 |
P6 |
0 |
3.0 |
0 |
10 |
2.12 |
2.12 |
1 |
�� |
�� |
P7 |
0.5 |
1.0 |
1.0 |
10 |
5.56 |
2.33 |
2.39 |
0.63 |
�� |
P8 |
0.5 |
0.5 |
1.5 |
10 |
5.9 |
2.12 |
2.78 |
0.79 |
2.68 |
P9 |
0 |
0.5 |
1.5 |
10 |
2.20 |
2.20 |
1 |
�� |
�� |
P10 |
0.5 |
0.5 |
1.5 |
10 |
5.88 |
2.25 |
2.61 |
0.70 |
�� |
P11 |
0.5 |
0.5 |
1.5 |
8 |
5.82 |
2.42 |
2.40 |
0.67 |
�� |
P12 |
0.5 |
0.5 |
1.5 |
12 |
5.23 |
1.98 |
2.64 |
0.68 |
�� |
2.2 Preparation of molecularly
imprinted monolithic column
The monolithic MIP was directly prepared by
in situ polymerization technique in a stainless steel chromatographic column of 125 mm��4.6
mm i.d. Template molecule (ENR, 0.5 mmol) was dissolved in methanol, to which the
functional monomer (see Table 1) was subsequently added. After stirring for 30 min, the
cross-linker (EDMA, 10 mmol), porogenic solvent (1-dodecanol, 2 mL) and free radical
initiator (AIBN, 0.08 mmol) were added stepwise. The mixture was degassed by
ultrasonication for 5 min, and deoxygenated by a stream of nitrogen gas for 5 min, and
then filled into a stainless steel column. The column was sealed and dipped into a 50�� water bath for 24 h. After polymerization, the
column was connected with a HPLC pump and washed exhaustively on-line with methanol-acetic
acid (9:1, v/v) to remove the template molecule and the porogenic solvent. Similarly, the
non-imprinted blank monolithic column (NIP monolith) without the imprinted molecule was
prepared in the same way for control experiments.
2.3 Chromatographic evaluation
A Shimadzu (Kyoto, Japan) LC-6A Liquid Chromatographic System with UV detector was used to
analyze the tested solutions. The UV detection wavelength was set at 280 nm. Eluent
flow-rate was 0.2 mL·min-1 to 1.0 mL·min-1 with
detection. The HPLC was carried out at the room temperature at a flow rate of 0.4 mL/min.
The 5 m L sample solution containing 1.0 mg/mL ENR in methanol was injected. Acetone was
injected as the void marker. Retention factor, k, was calculated by using the equation k =
(tR-t0)/t0, where tR and t0 are the
retention time of the analyte being investigated and the void marker, respectively. The
separation factor (a) is determined by the following equation, a = k2/k1,
where k1 and k2 are the retention factor of the ciproflocaxin and
the enroflocaxin, respectively. The resolution was calculated from the equation Rs = 2(t2
- t1)/(w1 + w2), where t1 and t2
are the retention time of the first and second eluted analytes, respectively, and w1
and w2 are the basedline peak widths of the first and second eluted analytes,
respectively. The imprinting factor (IF) was calculated from the equation IF = kimprinted/knon-imprinted.
2.4 Morphology analysis
After the chromatographic experiments had been completed, the column was washed with
methanol-acetic acid (9:1, v/v) for 4 h. The column fitting was removed and the monolithic
polymer was pushed out of the column using a flow-rate of 5 mL·min-1.
The polymer was dried under vacuum at 50�� for 24 h
and cut into pieces with a razor blade. Surface analysis of polymer was carried out in a
KYKY-2800B scanning electron microscopy (Beijing, China) at 25 kV.
2.5 Binding capacity and adsorption isotherms
Static method was performed by placing 50 mg polymer particles, 10.00 mL methanol and
different concentration of the template solution into 10 mL flasks, then the flasks were
oscillated in a constant temperature bath oscillator at 25 �� for 8 h. The mixture was transferred into a centrifuge tube and
centrifuged at 4000 rpm for 5 min. The concentrations of free compounds in the solutions
were determined by an UV-Spectrometer at 280 nm.
2.6 Determination of ENR in eggs
A sample of homogenised egg (5 g) was accurately weighed. In order to investigate the
effect of spiking procedure on extraction efficiency, the samples were spiked with the
mixture fluoroquinolone at 0.30, 0.40, 0.50 mg / g to each tube, respectively, add 3 mL acetonitrile and 0.25 mL
concentrated ammonium hydroxide, and homogenize. Centrifuge for 5 min at 3000 rpm, and
decant supernatant into new disposable 50 mL centrifuge tubes. The residue was washed with
water (1 mL)- acetonitrile (3 mL)- ammonium hydroxide (0.25 mL) and centrifuge. To each of
the combined supernatants, add 3 mL hexane, 3 mL ethyl ether, and 0.25 mL 1 M NaCl. Mix on
a Vortex mixer 15 s; remove and discard upper layer. Transfer lower layers into 18��150 mm
glass disposable test tubes, rinsing in with 4 mL acetonitrile. Evaporate samples at 40��
under a Vortex of nitrogen. Add additional 1 mL portions of
acetonitrile, as needed, to facilitate evaporation. Dissolve residues in 2 mL 0.1 M
phosphate, pH 9.0, and mix on a Vortex mixer 10 s. the sample solution was filtered
through a 0.45 m m membrane into amber autosampler vials for analysis. 10 m L of the
solution prepared was injected to the monolithic column directly.
3.1 Optimization of preparation conditions for the MIP monolithic column
The ENR imprinted monolith was prepared in chromatographic column using non-covalent imprinting technique. In order to acquire a monolith with high selectivity and low backpressure, some preparation conditions must be taken into account. Firstly, an appropriate monomer is the key factor for imprinting. In this study, the acidic (MAA), neutral (AM) and basic (4-VPY) compounds were investigated. The MAA and MMA were chosen as the functional monomer and the MIP monolith showed specific recognition ability to the template ENR, whereas the retention of its analogs is fairly weak. ENR and CIP molecule have one carboxyl group, but ENR molecule has one secondary amine group and CIP molecule has one primary amine group, therefore the acidic MAA and MMA are the best imprinting monomer for selective recognition.
In general, a proper template-monomer ratio can afford high selectivity. The influence of the template-monomer ratio (1:3, 1:4 and 1:6) on the selectivity was evaluated. As shown in Table 1, the ratio of 1:4 was optimal for the template molecule ENR, while higher or lower ratios provided lower selectivity. When the ratio was lower than 1:4, the MIP monolith columns had very weak retention for the template and its analogs. When the ratio was 1:6, the MIP monolith showed high cross-reactivity due to the relatively similar structures of ENR and CIP.
In the MIP technology, the amount of the cross-linker should be appropriate not only to maintain the stability of the recognition sites and the recognition cavities, but also to have some extent of the flexibility in order to make the template enter the cavities easily [2]. As shown in Table 1, when the molar ratio of monomer to cross-linker was 1:5, the monolithic MIP showed good affinity and selectivity for the template ENR. At higher crosslinking degree, the recognition for the template ENR decreased because it was more difficultf for the template to enter the cavities of the rigid MIPs. When the crosslinking degree is lower, the monolith showed high backpressure at high flow rate. The disadvantage of the shrinkage and tube wall adhesion need improved for MIP monolithic preparation in a stainless steel chromatographic column. In the study, copolymerization monomers were investigated in MIP monolithic preparation process. The influence of the MAA-MMA ratio (1:1, 1:3) on the selectivity was evaluated. As shown in Table 1, the ratio of 1:3 was optimal for the template molecule ENR, the imprinting factor was up to 2.68. The template molecules hydrophobic interact with MIP monolith were enhanced for selective separation ENR and CIP. The schematic model of the enrofloxacin imprinting mechanism is shown in Fig.2. This is in agreement with previous studies carried out by Nittsson [35] which have already proved that ionic interactions arised between the fatty amine and carboxylic acid, and hydrogen bonds generally exist between the carboxyls of the organic compound.
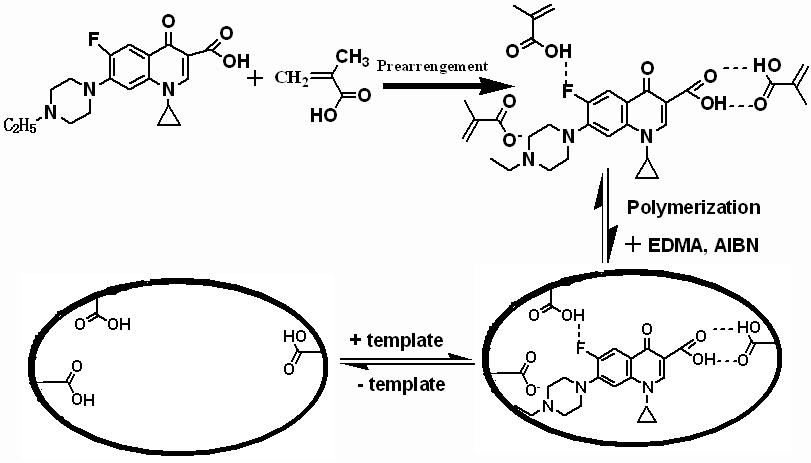
Fig.2 The schematic process of synthesized monolithic MIP
3.2 Effect of the mobile phase composition on the retention and resolution
The effect of the mobile phase on the retention and separation was investigated using methanol, chloroform and acetonitrile as mobile phase, respectively. The best separation was obtained by using chloroform as mobile phase. Enrofloxacin and ciprofloxacin were baseline separated on the MIP monolith, but no separation was observed on NIP monolith (Fig. 3). It indicates that molecular recognition was dependent on the stereo structures and the arrangement of functional groups of the imprinted molecule and the cavities on MIP.
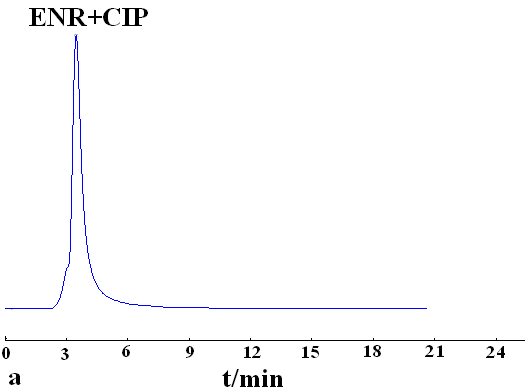
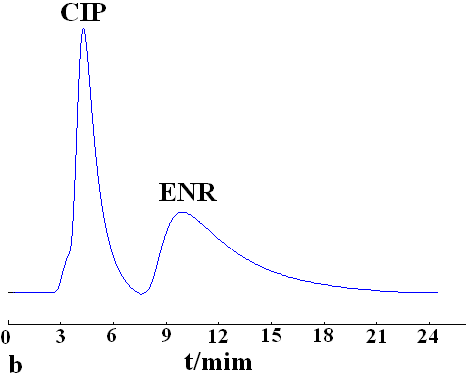
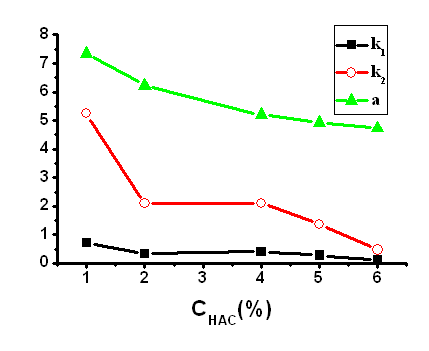
Mobile phase, chloroform-acetic acid; flow rate, 0.4 mL·min-1; detection wavelength, 280 nm; k1, the retention factor of ciprofloxacin; k2, the retention factor of enrofloxacin; ��, separation factor of ciprofloxacin and enrofloxacin.
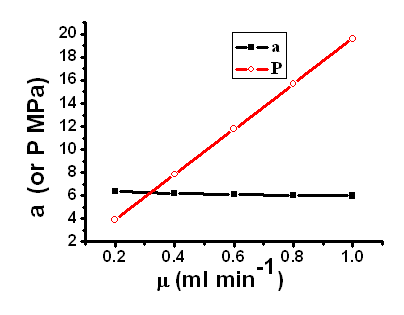 Fig.6 The relationship of separation factor or backpressure and flow rates on monolithic MIP column. Column: 125mm��4.6 mm I.D., mobile phase: chloroform-acetic acid(98:2, v/v). |

3.3 Effect of flow rate on the
separation
The SEM image (Fig.5.) of enrofloxacin
monolithic column showed that there were many macropores and flowthrough channels inlaid
in the network skeleton of enrofloxacin imprinted monolith. Through-pores provide flow
paths through the column, and the size and density of the macropore network gives the
monolithic columns a high external porosity and, consequently, a large permeability and
low column hydraulic resistance. At the same time, the network of mesopores is responsible
for the large specific surface area of the monolith. For these reasons, the monolithic MIP
columns are efficient at high flowrates and allow the achievement of very high
efficiencies. In this experiment, the flow rate of the mobile phase was investigated over
the range of 0.2-1.0 mL·min-1
(Fig.6.), and the results showed that although the migration times of the enrofloxacin and
ciprofloxacin decreased with increasing of the flow rate, merely a slight decrease of the
separation factorwas found with increasing flow rate. This is the typical characteristic
of monolithic column.
3.4 Binding capacity and adsorption isotherms
|
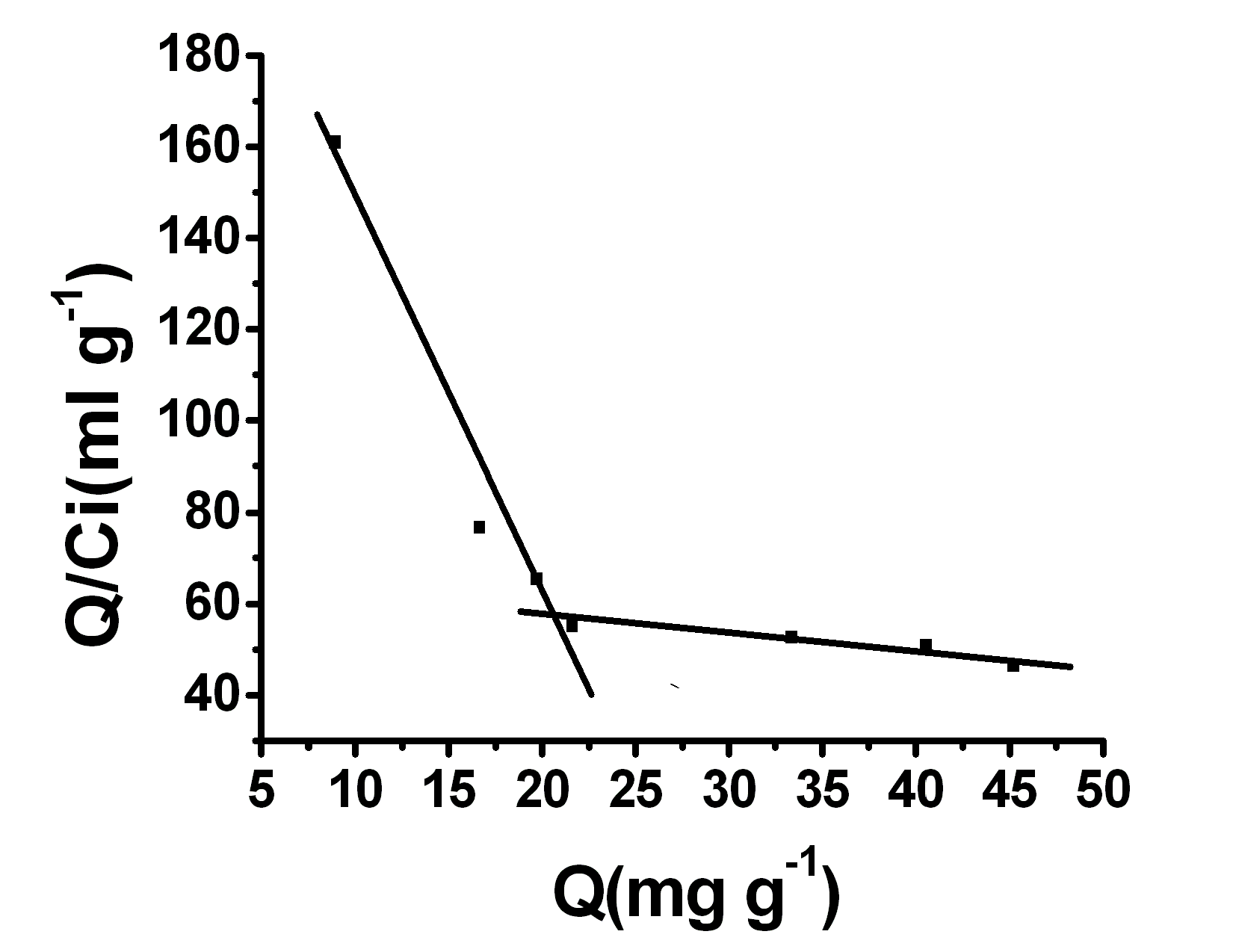
The binding capacities of the imprinted and non-imprinted monoliths were investigated by the Scatchard analysis mehtod (Fig.7.). The equilibrium binding experiments of the imprinted and non-imprinted polymers were carried out by varying the initial concentration of enrofloxacin in the range of 0 �C 1.2 mg·mL-1. The average binding data of triplicated independent results can be linearly transformed according to the Scatchard equation Q/C0 = (Qmax-Q)/KD, where KD is an equilibrium dissociation constant, Q is the binding capacity and Qmax is an apparent maximum binding capacity. When Q/ Cfree is plotted versus Q, KD and Qmax can be estimated from the slope and the intercept, respectively. The linear regression equations for the two linear regions are Q/ Cfree =231.70257-8.49582Q (r = 0.9813) and Q/ Cfree = 63.11093-0.33673Q (r = 0.9344).This suggests that there are two heterogeneous hydrogen bonds (H·· ·O and N·· ·H) in the MIP with specific binding properties and the affinity. From the slope (KD) and the intercept (Qmax) of the scatchard plot for the higher affinity binding sites can be calculated to be 0.3275 mmol·L-1 and 75.88 mmol·g-1 dry polymer, respectively. Similarly, the KD and Qmax for the lower affinity binding sites were found to be 8.264 mmol·L-1 and 521.5
mmol·g-1 dry polymer, respectively.3.5 Analysis of eggs
A calibration curve was obtained by loading 10 mL standard solutions of enrofloxacin over the range of (0.02-0.3 mg·mL-1). Good linearity was obtained in this concentration range (r = 0.999). The quantification limits for this compound was 80 ng L-1. The eggs samples were analyzed and had not found enrofloxacin and ciprofloxacin. The spiked egg samples was extracted according to section 2.6. All the samples were determined by standard addition method. The recoveries were calculated from the corresponding calibration curve and the results are shown in Table 2.
Table.2 The recoveries of ENR and CIP in eggs fortified with different concentrations
Sample |
Enrofloxacin |
Ciprofloxacin |
Recovery (%) |
|
Enrofloxacin |
Ciprofloxacin |
|||
1 |
0.3 |
0.3 |
115 |
95 |
2 |
0.4 |
0.4 |
132 |
83 |
3 |
0.5 |
0.5 |
107 |
91 |
Enrofloxacin imprinted monolithic columns were successfully designed, evaluated and applied for quantitative determination of ciprofloxacin and enrofloxacin in eggs. Ciprofloxacin and enrofloxacin were fully separated under this kind of monolithic MIP column and hydrogen bonding interaction and hydrophobic interaction played important roles in the retention and separation. The morphological characteristics of the monolithic MIP showed that both mesopores and macropores were formed in the monolith. The study results presented here have substantiated the significant research interest in monolithic MIP columns due to their ease of preparation, high separation efficiency, and rapid mass transport. Acknowledgment
This work was supported by National Science Foundation of China (No. 20675024), Natural Science Foundation of Hebei Educational Committee (No. 2006407), China Postdoctoral Science Foundation (No. 2005037629) and Science Foundation of Hebei University. REFERENCES
[1] Lu Y.K., Zhao N., Qin X.Y et al. Chem. J. Inter., 2008,10: 32.
[2] Lavignac N., Allender J.C.,. Brain K.R, Anal. Chim. Acta, 2004, 510:139.
[3] Yang H.H., Zhang S.Q., Tan F., et al. J. Am. Chem. Soc, 2005, 127:1378.
[4] Vlatakis G., Andersson L., Miller R. et al. Nature, 1993, 361: 645.
[5] Myriam D.A., Esther T., Antonio M.E. Anal. Bioanal. Chem., 2009, 393:899.
[6] Yan X., Zhou H.L., Zhang Z.L. et al. Spectrochim. Acta Part A, 2007, 66: 341.
[7] Lee W.C., Cheng C.H., Pan H.H., et al. Anal. Bioanal. Chem., 2008, 390: 1618.
[8] Haginaka J., Futagami A. J. chromatogr. A, 2008, 1185 (2):258.
[9] Huang X.D., Qin F., Chen X.M., et al. J. Chromatogr. B, 2004, 804: 13.
[10] Huang X.D., Zou H.F., Chen X.M., et al. J. Chromatogr. A, 2003, 984: 273.
[11] Yan H.Y., Tian M.L., Row K.H., J. Sep. Sci., 2008, 31: 3015.
[12] Yin J.F., Yang G.L., Che Y. n, J. Chromatogr. A, 2005, 1090: 68.
[13] Tamayo F.G., Turie E.l, Esteban A.M., J. Chromatogr. A, 2007, 1152: 32.
[14] Elena B.P., Sofia M., Guillermo O., et al. Anal. Bioanal. Chem., 2009, 393: 235.
[15] Prasad B.B., Sharma P.S., Lakshmi D., J. Chromatogr. A, 2007, 1173: 18.
[16] Brüggemann O., Visnjevski A., Burch R., et al. Anal. Chim. Acta, 2004, 504: 81.
[17] Kazuko H., Sakai Y., Kameoka K., et al. Sens. Actuators B-Chem., 2002, 86: 20.
[18] Dickert F.L., Lieberzeit P., Tortschanoff M.. Sens. Actuators B-Chem., 2000, 65: 186.
[19] Haginak J., Sanbe H., J. Chromatogr. A, 2001, 913: 141.
[20] Liu X., Chen Z.Y., Zhao R., et al. Talanta, 2007, 71: 1205.
[21] Qu G.R., Wu A.B., Shi X.Z., et al. Anal. Lett., 2008, 41: 1443.
[22] Ho K.C., Yeh W.M., Tung T.S., et al. Anal. Chim. Acta, 2005, 542: 90.
[23] Wang J.F., Cormack P.A.G., Sherrington D.C., et al. Angew. Chem. Inter. Ed., 2003, 42: 5336.
[24] Han D.M., Fang G.Z., Yan X.P., J.Chromatogr. A, 2005, 1100: 131.
[25] Altmaier S., Cabrera K., J. Sep. Sci, 2008, 31: 2551.
[26] Matsui J., Kato T., Takeuchi T., et al. Anal. Chem., 1993, 65: 2223.
[27] Ou J.J., Kong L., Pan C.S., et al. J. Chromatogr. A, 2006, 1117: 163.
[28] Franco E.J., Hofstetter H., Hofstetter O., J. Pharm. Biomed. Anal., 2008, 46: 907.
[29] Matsui J., Miyoshi Y., Matsui R., et al. Anal. Sci., 1995, 11: 1017.
[30] Schweitz L., Spégel P., Nilsson S., Analyst, 2000, 125: 1899.
[31] Schweitz L., Andersson L.I., Nilsson S., Anal. Chim. Acta, 2001, 435: 43.
[32] Toussaint B., Chedin M., Bordin G., et al. J. Chromatogr. A, 2005, 1088: 32.
[33] Hassouan M. K., Ballesteros O., Zafra A., et al. J. Chromatogr. B, 2007, 859: 282.
[34] Hassouan M., Ballesteros O., Vlchez J., et al. Anal. Lett., 2007, 40: 779.
[35] Yan, H., Row, K.H. Biomed. Chromatogr., 2008, 22: 487.
[36] Sun H.W., Qiao F.X.. J.Chromatogr. A, 2008, 1212: 1.
[37] Liu H., Zhuang X., Turson M., et al. J. Sep. Sci., 2008, 31: 1694.
[38] Caro E., MarcéR.M., Cormack P.A.G., et al. Anal. Chim. Acta, 2006, 562: 145. ��ŵɳ�Ƿ���ӡ��Һ��ɫ�����������Ʊ������ڼ�⼦������ҩ������Ӧ��
���˿�*���� Խ���� ����½����������Ӣ
���ӱ���ѧ��ѧ�뻷����ѧѧԺ���ӱ�ʡ������ѧ�����ص�ʵ���ң��ӱ� ���� 071002��
ժҪ �Զ�ŵɳ��Ϊģ����Ӳ���ԭλ���۷������Ʊ����ŵͪ��ҩ�����ӡ�������������������ϵĵ羵��Ƭ��ʾ�����������ϵ���״�Ǽܽṹ��������ͨ���ʹ����ף���Щ�����ڽ��ʹ������������ֵ���ѹ���Ż���ɫ�������������˷���ӡ���ۺ������ʶ�������ͨ��Scatchard�������ֱ�����˾ۺ���߽��λ��Զ�ŵɳ�ǵ���ⳣ��KD1 (0.3275 mmol L-1)�ͱ�����������Qmax1 (75.88 ��mol g-1)���ͽ��λ�����ⳣ��KD2 (8.264 mmol L-1)�ͱ�����������Qmax2 (521.5 ��mol g-1)����������ȣ��÷���ֱ���Ʊ��˿����ȵķ���ӡ���������������˺�ʱ�ķ��顢��ĥ����ɸ�IJ��裬�����ӡ���ۺ��������ѡ���ԣ��ü����ɹ���Ӧ���ڼ����л���ɳ�����ŵɳ�ǵ�ѡ���Է��������
�ؼ��� ����ӡ���ۺ������������ŵɳ�ǣ�ʳƷ ��