Milan Kurfürst, Pavel Pihera and Jiří Svoboda*
Department of Organic Chemistry, Institute of Chemical Technology, Technicka 5, CZ-166 28 Prague 6, Czech Republic
Tel. ++-420-2-243101877, fax ++-420-2-2435 4288, E-mail: [email protected]
Abstract: A different reactivity of the title compounds in electrocyclic reactions with various dienophiles was found. While 2-vinyl[1]lbenzothieno[3,2-b]furan reacted as a diene, 2-vinylthieno[3,2-b][1]benzofuran due to its reduced diene conjugation showed also reactivity of an alkene in [2π+2π] mode. The obtained results support the hypothesis of a different aromaticity of both isomeric heterocyclic systems.
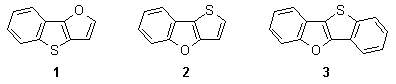
Benzoannelated thieno[3,2-b]furans belong to the family of A,B-diheteropentalenes.1 Due to our interest in the chemistry of annelated heterocyclic systems2-8, we studied recently synthesis, reactivity and material applications9,10 of these compounds. It was shown that stability and reactivity in electrophilic substitution and electrocyclic reactions of [1]benzothieno[3,2-b]furan (1) is different from its isomer, thieno[3,2-b][1]benzofuran (2).
Heterocycle 1 reacted5 as a dienophile with various substituted buta-1,3-dienes to create the corresponding cycloadducts 4 (some representative examples are shown in Scheme 1) that could be easily oxidized to a new dibenzo fused system 3 possessing a substituent in the benzofuran part of the molecule. Heterocycle 2 did not enter such cycloaddition reactions and starting compound was always recovered.
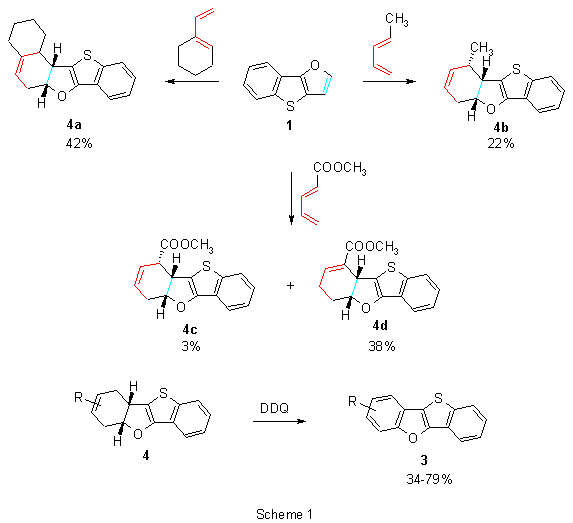
From the results it was emphasized that aromaticity of both isomeric heterocyclic is different and it is governed by the most stable heterocyclic moiety in the molecule. In the π-electronic system of 1, aromaticity is presumably controlled by the benzo[b]thiophene unit and structure of 1 can be considered to be a substited benzo[b]thiophene with an attached enol-ether moiety. The double bond then enters separate cycloaddition reactions as an electron-poor dienophile. On the other hand, aromaticity of 2 is governed by the stable thiophene ring and the molecule can be considered to be a phenyl (or phenoxy) substituted thiophene. This hypothesis was also supported by ab initio calculations of heats of hydrogenation of diheteropentalenes.11
We tried to support our assumptions by other experiments. We changed the dienophilic system in 1 and 2 to a diene by introduction of a double bond in position 2 and studied reactivity of these new systems in a series of electrocyclic reactions with reactive dienophiles.
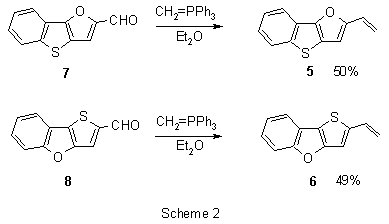
The starting 2-vinyl[1]lbenzothieno[3,2-b]furan (5) and 2-vinylthieno[3,2-b]-[1]benzofuran (6) were obtained by Wittig reaction of aldehydes 7 and 8 with methylene triphenyl phosphorane in a moderate yield.
To study cycloaddition reactions6, we chose methyl propiolate, maleic anhydride, acrylonitrile and dimethyl acetylenedicarboxylate (DMAD) as suitable and reactive dienophiles. Heating of methyl propiolate with vinyl derivative 5 in benzene for 32 h led to a sole and fully aromatized product 9. Reaction with maleic anhydride proceeded smoothly and the corresponding cycloadduct 10 was obtained in a good yield. Due to low boiling point of acrylonitrile, cycloaddition was performed in a sealed tube at 125 °C and from the reaction mixture adduct 11 was isolated (Scheme 3).
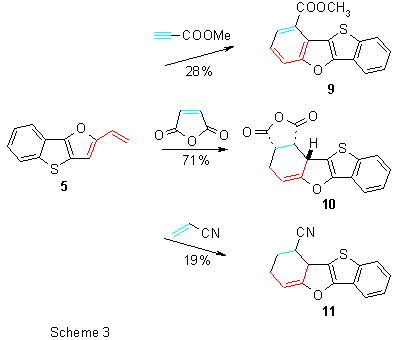
Because no other regio- and stereoisomers were obtained, it can be emphasized that the cycloaddition reaction proceeded regiospecifically and with endo-selectivity (compound 10).
Reaction of 5 with DMAD proceeded, however, in a more complicated way. Heating of both components in toluene afforded a mixture of two products - diester 12 and the major triester 13. From the structure of 13, it can be concluded that a series of consecutive transformations took place (Scheme 4): rearrangement of the double bond in the primary adduct 14 into conjugation with the second double bond creates a new diene 15. Its cycloaddition with another molecule of DMAD leads to a bridged adduct 16 which in a subsequent retro-Diels-Alder reaction loses methyl acrylate and forms the major product 13. None of the intermediates 14-16 could be detected by tlc. Presence of methyl acrylate was qualitatively proved by GC with an authentic sample.
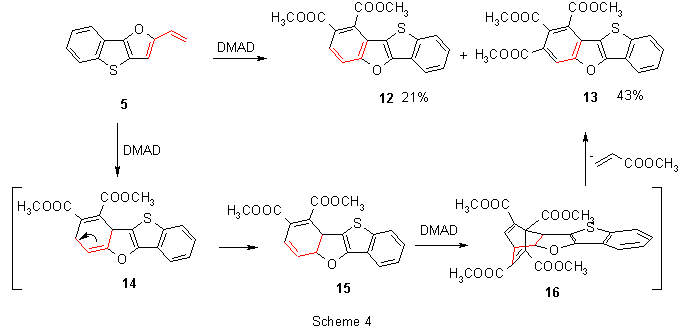
The isomeric vinyl derivative 6 showed in comparison with 5 much lower reactivity in Diels-Alder cycloaddition reactions. Only after a long-term heating of 6 with methyl propiolate in toluene the corresponding fully aromatized ester 17 was obtained in a low yield (Scheme 5).
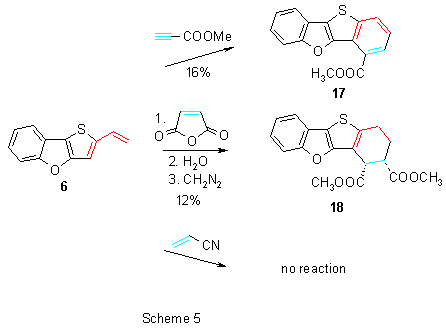
Cycloaddition reaction of 6 with maleic anhydride proceeded also very slowly. Because the primary adduct was not stable, the crude reaction mixture was hydrolyzed and treated with diazomethane. Diester 18 was then obtained by column chromatography. Reaction of 6 with acrylonitrile under conditions given above did not proceed at all and no product of a [4π+2π]-cycloddition reaction was detected.
Analogously to vinyl derivative 5, we expected an more complex course of reaction of 6 with DMAD. To our surprise, after a long-term heating (58 h) of both components in toluene we obtained a mixture of five products that were separated by a thorough column chromatography (Scheme 6).
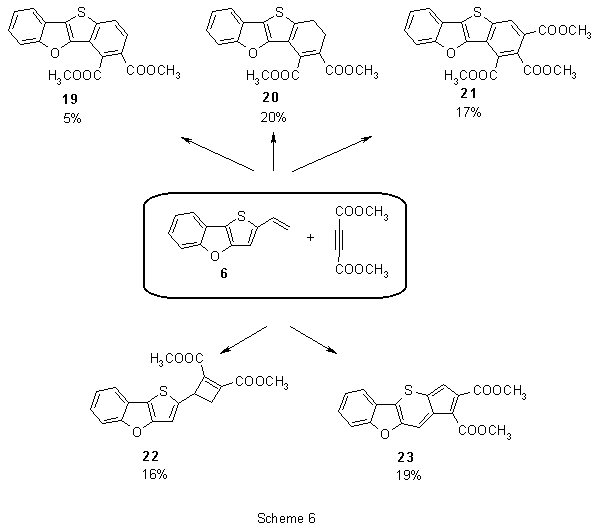
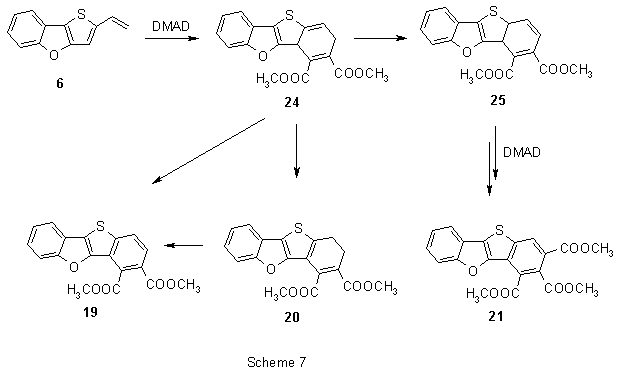
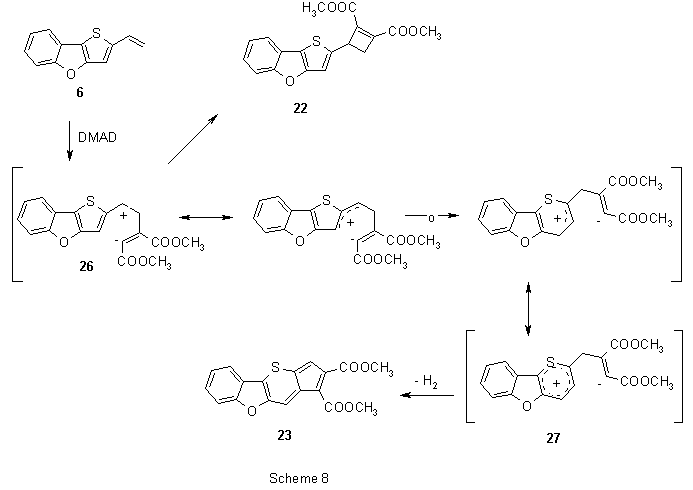
In addition to the expected products - diester 19 (5% yield) and triester 21 (17%), also dihydroderivative 20 was isolated (20%). All the products resulted from a [4π+2π]-cycloaddition reaction. However, presumably due to reduced conjugation of the dienic system, the vinyl group entered a separate reaction with DMAD leading to two products of [2π+2π]-cycloaddition: cyclobutene derivative 22 (16%) and intensively cyan-coloured product 23 (19%) were obtained.
The primary adduct 24 (Scheme 7) of [4π+2π]-cycloaddition can obviously rearrange the exocyclic double bond (relative to thiophene) to form the dihydro derivative 20 or to create a new dienic system in 25. This diene reacts with DMAD and in a series of consecutive reactions analogous to that in Scheme 4 triester 21 is formed. Dehydrogenation of the primary adduct 24 and 20 leads to formation of diester 19.
Another and no less important direction of transformation of 6 represents formation of products 22 and 23. Although thermic [2π+2π]-cycloadditions are symmetrically forbidden, non-concerted processes leading to formation of a four-membered ring derivatives have been described12 through diradical and zwitterionic intermediates, resp., or charge-transfer complexes. In analogy with other reactions of DMAD13-15, we assume that the reaction also proceeds through zwitterionic intermediates. Addition of electron-rich vinyl group to extremely electron-poor triple bond of DMAD creates a zwitterionic intermediate 26 (Scheme 8) that may close the cyclobutene ring and produce 22. Stability of zwitterion 26 can be increased by π-electrons overlap of the neighbouring thiophene ring that is followed by ring expansion of thiophene to thiopyrane and formation of a new zwitterionic structure 27. The new formed cation can be stabilized analogously by the thiopyrane ring. After closure the cyclopentane ring and dehydrogenation, compound 23 is formed. Although fused cyclopentathiopyranes as analogues of azulenes have been reported16-21, they have never been obtained by such a ring enlargement. In addition, only few examples of thiopyrane synthesis starting with S,C-ylides (thiophenium ylides) are known22,23. Structure of the cyclobutene derivative 22 was assigned unambiguously by 1H, 13C NMR spectra and correlation experiments. However, we were not able to assign the structure of compound 23 by these methods. Fortunately, a single crystal of this substance was obtained and the structure could be then established by X-ray crystallography.
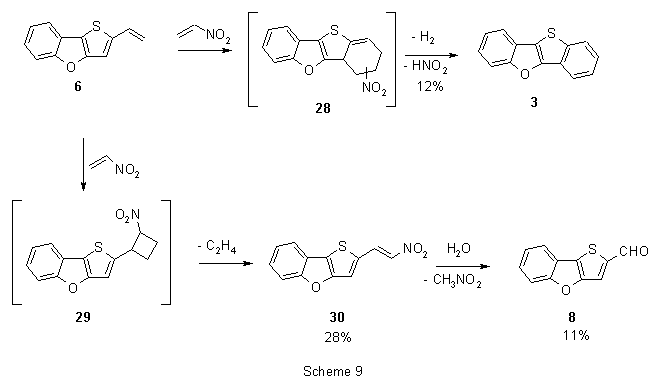
A competitive [2π+2π]-cycloadditions was often observed with Diels-Alder reactions with extremely electron-poor dienophiles12. To obtain further information of this phenomenon, we studied cycloaddition reaction of 6 with nitroethylene. Heating of both substances in benzene for 38 h afforded a mixture of three product that were separated by column chromatography. The minor product (12% yield) was identified to be the parent compound 3 (Scheme 9). Major products were nitrovinyl derivative 30 and aldehyde 8. We assume that heterocycle 3 is formed by aromatization of the primary [4π+2π]-adduct by dehydrogenation and coincident loss of nitrous acid24,25. To explain the formation of major products, we suppose that prevailing competitive [2π+2π]-cycloaddition takes place and cyclobutane 29 is formed. In the subsequent step, retro-ring opening with the loss of ethylene affords nitro derivative 30 that in the presence of a trace of water and by aqueous workup, resp., leads to formation of aldehyde 31. However, neither intermediates 28 nor 29 could be detected during the reaction by tlc. Structure of 30 was also confirmed in an independent attempt: reaction of aldehyde 8 with nitromethane in the presence of potassium carbonate afforded compound 30. We consider the presented mechanism to be more likely than other eventualities:
Vinyl derivatives 5 and 6 show different patterns of reactivity in studied cycloaddition reactions. Reactivity of the former derivative resembles behaviour of 2-vinylbenzo[b]furan and 2-vinylfuran. Diels-Alder reactions of 5 can be practically used for synthesis of tetracyclic derivatives of 3 possessing substituents in the "benzofuran" part of the molecule. When electron-poor and very reactive DMAD was used, cycloaddition reaction proceeded by a more complicated course. Cycloadditions of vinyl derivative 6 with common dienophiles proceeded slowly. In spite of this fact, cycloaddition enables to prepare derivatives of 3 possessing a substituent in the "benzothiophene" part. Application of extremely electron-poor dienophiles (DMAD, nitroethylene) leads to a substantial change in the course of cycloaddition. Because reduced conjugation of the vinyl double bond with the 2,3-double bond in 6 leads to slowdown of the [4π+2π]-process, competitive [2π+2π]-cycloaddition becomes the important route of transformation. Change in the course of reaction may result from the change of kinetic and thermodynamic controll of cycloaddition reaction12.
Results of this study confirm our concept of a different aromaticity of isomeric heterocycles 1 and 2.
Financial support from the Grant Agency of Czech Republic, Project. No. 106/00/0580, is gratefully acknowledged.