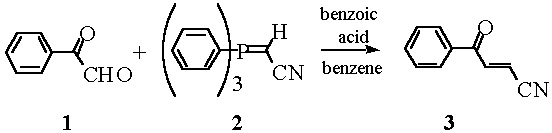
| Molbank 2003, M334 |
3-Benzoylacrylonitrile [(E)-4-Oxo-4-phenyl-2-butenenitrile]
Thies Thiemann* and Masataka Watanabe
Institute of Advanced Material Study and Graduate School of Engineering Sciences, Kyushu University, 6-1, Kasuga-koh-en, Kasuga-shi 816-8580, Japan
Fax: 0081-92-583-7811, E-mail: [email protected]
Received: 18 February 2003 / Accepted 22 May 2003 / Published 1 June 2003
3-Benzoylacrylonitrile (3) has been prepared previously. Its preparations [1-4] however, are usually multistep reactions, and most of the sequences necessitate the use of KCN. There has been a report that phenylglyoxal 2,2-dimethylacetal can be reacted with cyanomethylidenetriphenylphosphorane [5,6]. In the following, it is noted that the commercially available phenylglyoxal monohydrate can also be used directly in a Wittig reaction with 2 as 2, a stabilized Wittig reagent, reacts with the carbaldehyde function much more readily than with the keto group. Important for the success of the reaction is the azeotropic removal of the water from phenylglyoxal monohydrate before the addition of the Wittig reagent. 3-Benzoylacrylonitrile (3) has been used as key intermediate for the construction of heterocycles [7] and has been reacted as a Michael acceptor [8,9] to furnish 3-aminopropiophenones of pharmaceutical value [9].
A solution of phenylglyoxal monohydrate (720 mg, 4.74 mmol) in benzene (20 mL) was heated under reflux with azeotropic removal of water (Dean Stark water collector filled with MS 4 Å). Thereafter, cyanomethylidenetriphenylphosphorane [10] (1.80 g, 5.98 mmol) and benzoic acid (100 mg, 0.82 mmol) were added and the resulting mixture was stirred for 10 h under an argon atmosphere at 80°C. Then, additional 2 (900 mg, 2.99 mmol) and benzoic acid (50 mg, 0.41 mmol) were added and the reaction continued at 80°C for a further 4 h. The cooled mixture was concentrated in vacuo and the residue was subjected to column chromatography on silica gel (hexane/ether 2:1) to give 3 as colorless needles (510 mg, 68%); mp 80°C.
IR (KBr) n 3038, 2220, 1665, 1604, 1592, 1446, 1339, 1273, 959, 690 cm-1.
1H NMR (270 MHz, CDCl3) d 6.59 (d, 1H, 3J 15.9 Hz), 7.55 (m, 2H), 7.67 (m, 1H), 7.82 (d, 1H, 3J 15.9 Hz), 7.98 (d, 2H, 3J 7.3 Hz).
13C NMR (67.9 MHz, CDCl3) d 111.97, 116.33, 128.82, 129.15, 134.54, 135.49, 141.58, 186.39.
MS (70 eV) m/z (%) 157 (68), 129 (28), 105 (100), 77 (43).
References and Notes:
[1] Hosokawa, T.; Aoki, S.; Murahashi, S. Synthesis 1992, 558.
[2] Nudelman, A.; Keinan, E. Synthesis 1982, 687.
[3] Severin, T.; Poehlmann, H. Chem. Ber. 1978, 111, 1564.
[4] Nesmeyanov, A. N.; Rybinskaya, M. I. Dokl. Akad. Nauk SSSR 1957, 115, 315; Chem. Abstr. 1958, 52, 7158g.
[5] Raulet, C. Compt. Rend. Sci. Acad. Sci., Ser. C 1973, 276, 903.
[6] Raulet C. Bull. Soc. Chim. Fr. 1974, 531.
[7] Akiyama, Y.; Abe, J.; Takano, T.; Kawasaki, T.; Sakamoto, M. Chem. Pharm. Bull. 1984, 32, 2821.
[8] Colonna, M.; Marchetti, L. Gazz. Chim. Ital. 1966, 96, 1175.
[9] Ger. Offenl. DE 2.116.293 (Inv.: Raabe, T.; Stachel, A.; Schottholt, J.; Nitz, R. E.; Casella Farbwerke Mainkur A.-G.; 19.10.1972 [Appl. 2.4.1971]); Chem. Abstr. 1973, 78, 16219.
[10] Trippett, S.; Walker, D.M. J. Chem. Soc. 1959, 3874.
Sample Availability: Available from the authors
© 2003 MDPI. All rights reserved.