| Molbank 2007, M555 |
Keywords: 7-Deazaadenine, Tubercidin, Functionalization
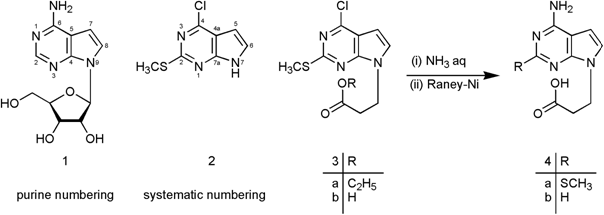
The adenine-isosteric 7-deazaadenine represents the heterocyclic nucleobase of the nucleoside antibiotic tubercidin (1) [1]. This base is able to form regular Watson-Crick but not Hoogsteen base pairs [2].
Functionalized derivatives of 7-deazaadenine such as compound 4b are of considerable interest because they can be easily coupled to polymers or surfaces carrying amino functions lending them the functionality of a particular modified nucleobase [3]. Coupling of compounds 3b or 4a,b to amino-functionalized lipids [4] or phospholipids such as kephalines may lead to potential organo- or hydrogelators. Moreover, compound 4b represents the 7-deaza analogue of 3-(adenine-9-yl)propanoic acid – a by-product of the antihypocholesteremic eritadenine isolated from the Shiitake mushroom Lentinus edodes Sing [5]. Furthermore, compounds 3b and 4a,b can be used for the synthesis of base-modified peptidyl nucleic acids (PNA) [6].
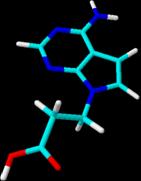
The preparation of 4b started from 4-chloro-2-(methylthio)-7H-pyrrolo[2,3-d]pyrimidine (2) – synthesized in 5 steps according to the literature [7-10] - which was alkylated with ethyl 3-bromopropionate under liquid-liquid phase transfer catalysis conditions with tetrabutylammonium hydrogensulfate as catalyst to yield compound 3a [3]. Subsequent saponification of the ester group gave the acid 3b. Nucleophilic displacement of the 4-chloro substituent by an amino group using aqueous ammonia (→ 4a) followed by reductive removal of the 2-SCH3 group with Raney Nickel gave the title compound 4b; its 3D-optimized structure is shown in the figure. Studies on the coupling of 4b to various carrier molecules is underway.
Experimental
Procedures
General
Thin-layer chromatography (TLC): Silica Gel 60 F254 plates (VWR, Darmstadt, Germany). UV Spectroscopy: U-3200 spectrophotometer (Hitachi, Japan) λmax in nm, ε in dm3/mol. NMR Spectra were measured on AC-250 and AMX-500 spectrometers (Bruker, Rheinstetten , Germany). Operational frequencies: 1H: 250.13, 500.14 MHz; 13C: 62.896, 125.700 MHz. Chemical shifts (δ values) are in parts per million relative to tetramethylsilane as internal standard. Microanalyses were performed by Mikroanalytisches Labor Beller (Göttingen, Germany). The 3D-optimized structure of compound 4b shown in the figure was obtained using the program ChemSketch/3D Viewer, version 10.0, from Advanced Chemistry Developments, Toronto, Canada; http://www.acdlabs.com.
3-[4-Chloro-2-(methylthio)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]propanoic acid (3b)
4-Chloro-2-(methylthio)-7H-pyrrolo[2,3-d]pyrimidine
(2,
0.5 g, 2.5 mmol), suspended in a mixture
of 20 ml of benzene and 20 ml of 50% aq sodium hydroxide, and
tetrabutylammonium hydrogensulfate (85 mg, 0.25 mmol) were agitated for
5 min
with a vibromixer. Thereupon, ethyl 3-bromopropionate (3.11 ml, 25
mmol) was
added and mixing was continued for 60 min. Then, another portion of
ethyl
3-bromopropionate (3.11 ml, 25 mmol) was added. After further mixing
for 30 min
the layers were separated and the aqueous phase extracted twice with
benzene.
The combined organic layers were washed with water, filtered and
evaporated to
dryness. The residue was dissolved in a small amount of MeOH, and ethyl
3-[4-chloro-2-(methylthio))-7H-pyrrolo[2,3-d]pyrimidin-7-yl]propanoate
(3a) was crystallized by addition of water.
Colorless crystals (523 mg,
69%); m.p. 62-63°C. TLC (silica gel, CHCl3-MeOH,
9:1, v/v): Rf
0.9. UV (MeOH): λmax 252, 279, 309 nm (ε,
26.750, 5.600, 6.500). Anal. calcd. for C12H14N3O2SCl
(299.785): C, 48.08; H, 4.71; N, 14.02. Found: C, 48.24, H, 4.81; N,
13.97. 1H-NMR
(d6-DMSO): δ, 1.09 (3H, t, CH3-ester,
J
= 7.0 Hz); 2.59 (3H, s, SCH3);
2.93 (2H, t, CH2N, J =
7.0 Hz); 4.01 (2H, q, CH2-ester, J
= 7 Hz); 4.47 (2H, t, CH2C=O, J
= 4.0 Hz); 6.53 (1H, d, H-C(5), J
= 4.0 Hz); 7.59 (1H, d, H-C(6), J
= 4.0 Hz). 13C-NMR (d6-DMSO):
δ, 13.8 (CH3-ester);
13.9 (SCH3); 34.0 (CH2C=O);
39.1 (CH2-ester);
60.2 (CH2N); 98.7 (C-5); 113.5 (C-4a); 129.8
(C-6); 150.6 (C-7a);
151.4 (C-2); 163.0 (C-4); 170.5 (C=O). The ester 3a (200
mg, 0.67 mmol) was dissolved in a
mixture of 10 ml of EtOH and 10 ml of 1N NaOH and stirred for 30 min at
room
temperature. After dilution with 50 ml of water the reaction mixture
was
neutralized by addition of Amberlite IR-120 (H+-form,
glass
electrode). After filtration the ion exchange resin was thoroughly
washed with
EtOH/H2O (1:1, v/v). The filtrate was evaporated
to dryness, and the
residue was taken up in a small amount of water. The title compound (3b) was
crystallized by adding a few drops
of glacial acetic acid. Colorless crystals (175 mg, 96%); m.p.
147-148°C. TLC
(silica gel, CHCl3-MeOH, 9:1, v/v): Rf
0.3. UV (MeOH): λmax 252, 279, 309 nm (ε, 26.300, 5.500, 6.000).
Anal. calcd. for C10H10N3O2SCl
(271.731): C, 44.20; H, 3.71; 15.47. Found: C, 44.36; H, 3.85; N,
15.29. 1H-NMR
(d6-DMSO): δ, 2.59 (3H, s, SCH3);
2.95 (2H, t, CH2N,
J
= 7.0 Hz); 4.47 2H, t, CH2C=O,
J
= 7.0 Hz); 6.53 (1H, d, H-C(5),
J
= 4.0 Hz); 7.58 (1H, d, H-C(6),
J
= 3.8 Hz). 13C-NMR (d6-DMDO):
δ, 13.7 (SCH3); 33.7 (CH2C=O);
51.4 (CH2N);
98.7 (C-5); 113.5 (C-4a); 129.8 (C-6); 150.6 (C-7a); 151.6 (C-2); 163.0
(C-4);
170 9 (C=O).
3-(4-Amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)propanoic
acid (4b)
The
acid 3b
(300
mg, 1.1 mmol) was dissolved in
conc. aq. ammonia (25%) and heated to 120°C in an autoclave for 12 h.
After
evaporation to dryness the residue was dissolved in dilute aq. ammonia,
and the
solution was acidified by addition of glacial acetic acid. Colorless
needles of
compound 4a
(183 mg, 66%); m.p.
267-270°C. TLC (silica gel, 0.25 M LiCl): Rf
0.5. UV
(MeOH): λmax 239, 284 nm (ε, 17.500, 12.200). Anal. calcd.
for C10H12N4O2S
(252.30): C, 47.61; H, 4.79; N, 22.21. Found: C, 47.60; H, 4.95; N,
22.34. 1H-NMR
(d6-DMSO): δ, 2.45 (3H, t, SCH3);
2.74 (2H, t, CH2N,
J
= 7.0 Hz); 4.26 (2H, t, CH2C=O,
J
= 6.9 Hz); 6.43 (1H, d, H-C(5),
J
= 3.4 Hz); 6.99 (1H, d, H-C(6),
J
= 3.5 Hz); 7.01 (2H, s, NH2).
13C-NMR (d6-DMSO): δ,
13.0 (SCH3); 34.4, 40.0
(2 x CH2); 98.4 (C-5); 99.6 (C-4a); 122.7 (C-6);
150.1 (C-7a); 156.8
(C-4); 162.4 (C-2); 172.0 (C=O). The
acid 4a
(300 mg, 1.19 mmol) was dissolved in 25%
aq. ammonia (50 ml), and Raney
Nickel
suspension (2 ml) was added. After refluxing for 2 h, the catalyst was
filtered
off, thoroughly washed with hot aq. ammonia, and the filtrate was
evaporated to
dryness. The residue was dissolved in dilute aq. ammonia and
chromatographed on
Dowex 1x2 ion exchange resin (OAc- form; column:
2.5 x 40 cm).
By-products were eluted first by water; the title compound was eluted
by dilute
acetic acid (10 vol-%). Colorless crystals of compound 4b (182
mg, 74%); m.p. 282-285°C. TLC
(silica gel, 0.25 M LiCl): Rf 0.55. UV
(MeOH, MeOH-H2O, 1:1 v/v): λmax 272 nm (ε, 7.300). Anal. calcd. for C9H10N4O2
(206.20): C, 52.42; H, 4.89; N, 27.17. Found: C, 52. 47; H, 4.99; N,
27.12. 1H-NMR
d6-DMSO): δ, 8.05 (1H, s, H-C(2)); 7.12 (1H, d,
H-C(6), J
= 3.4 Hz); 6.95 (2H, s, NH2);
6.49 (1H, d, H-C(5), J
= 3.4 Hz);
4.31 and 2.75 (2 x 2H, 2 t, 2 x CH2, J = 7.0
Hz, both). 13C-NMR (d6-DMSO): δ,
171.9 (C=O); 157.2 (C-4); 151.2 (C-2); 149.3 (C-7a); 123.9 (C-6);
102.3 (C-4a); 98.1 (C-5); 40.0 and 34.3 (2 x CH2).