
Asymmetric Synthesis
on Solid Support
using Polymer-bound Auxiliaries [1]
Kekulé-Institut
für Organische Chemie und Biochemie der Rheinischen Friedrich-Wilhelms-Universität
Bonn, Gerhard-Domagk-Str. 1
Institut für
Organische Chemie, D-53121 Bonn, Germany, Telefax:
Int. +
(49) 228/739712, E-mail:
[email protected]
Received: 27 July 2001 / Uploaded 7 August 2001
Abstract
Introduction
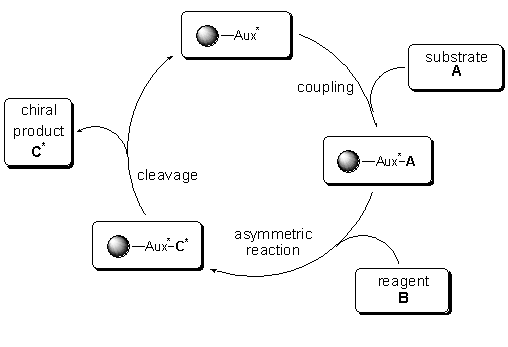
Figure 1
Recycling scheme for a polymer supported auxiliary
Recycling
may be the key issue, besides from easy workup procedures, for the use
of polymer-bound chiral auxiliaries. The employment of chiral auxiliaries
in industrial drug discovery processes is limited, due to their price and
availability. Reusable auxiliaries on solid support might overcome this
drawback. Especially in the high-throughput approaches of compound libraries
using solid-phase synthesis, the use of chiral linkers might have an additional
impact.
Polymer-bound Auxiliaries
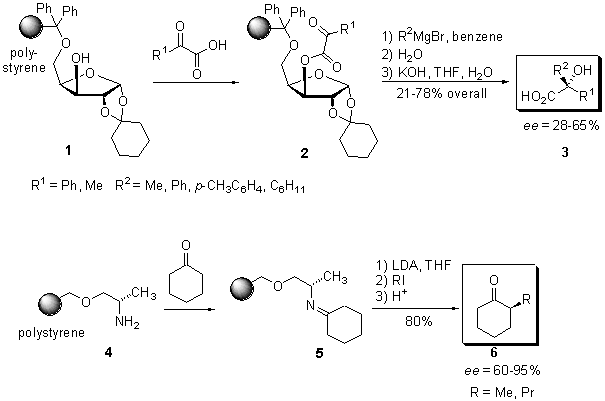
Scheme
1 The two first polymer-bound chiral auxiliaries in solid-phase chemistry
In
1979 Leznoff et al. (3) reported
the synthesis of an immobilized alaninol-auxiliary and its application
in the asymmetric a-alkylation
of cyclohexanone (Scheme 1). The polymer-bound auxiliary 4, readily
prepared by the attachment of N-phthalimide protected alaninol to
Merrifield resin under basic conditions and subsequent deprotection, was
converted with cyclohexane to its corresponding imine 5. After deprotonation
with lithium diisopropylamide (LDA), alkylation with iodo methane
or propane at room temperature and acidic hydrolysis, the 2-alkylcyclohexanones
6
were obtained in good yields (80%) and good to excellent enantiomeric excesses
(60 and 95%, respectively). The achieved enantiomeric excess of the products
was as least as high as for the synthesis in solution. Again, the polymer-bound
auxiliary 4 could be reused without any loss of enantioselectivity.
In
1981, the synthesis of a-alkylated
esters using a polymer-bound Meyers oxazoline was demonstrated by the group
of McManus (4). A chiral oxazoline,
derived from hydroxy phenyl alaninol and attached to Merrifield resin,
was alkylated with benzyl chloride to provide, after acid catalysed ethanolysis, a-alkylated
propionic acid ester. Due to incomplete cleavage from the solid support
the chemical yield was poor (43-48%). In addition, the optical purity of
the product (ee = 56%) was much lower as reported for the same reaction
in solution phase.
These early results clearly showed that asymmetric synthesis on solid support and recycling of the polymer-bound chiral auxiliary is possible. However, up to the early nineties of the last century, no further investigations in this field were done. From that time on, several immobilized auxiliaries, especially Evans oxazolidinones and proline-type auxiliaries, were developed and applied to different reactions.
Kurth
and coworkers (5) reported the solid-phase
synthesis of 3,5-di-substituted g-butyrolactones
by iodolactonisation of immobilized (S)?prolinol amides. In the
first step, N-acylated (S)-prolinol was attached to Merrifield
resin under basic conditions followed by deprotonation of the obtained
resin 7 with LDA and subsequent addition of methyliodide to furnish
polymer-bound Ca-methylated
amide 8 (Scheme 2).
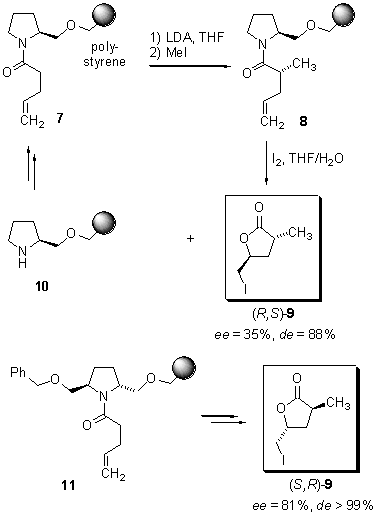
Scheme 2 Iodolactonisation
of polymer-bound alkenes
Formation
of the g-butyrolactone
and cleavage from the solid support was achieved by stirring a THF/water
mixture of resin 8 with iodine for three days. After separation
of the resin bound (S)-prolinol 10 by filtration, g-butyrolactone
(S,R)-9 was obtained in 33% overall yield as a 64:4:31:2
mixture of isomers, representing a 94:6 trans versus cis
selectivity and an enantiomeric excess of ee = 35%. The authors
were also able to demonstrate the recovery and recyclisation of the chiral
auxiliary through the reaction sequence.
This
methodology was extended by the development of a second-generation pseudo-C2-symmetric
pyrrolidine-based auxiliary 11 (Scheme 2) (6).
Following the procedure described above, the (R)-configured auxiliary
11
furnished g-butyrolactone
(R,S)-9 in 34% overall yield as a single diastereomer
with an enantiomeric excess of ee = 81%. In contrast to the (S)-prolinol
derived auxiliary 10, the pseudo-C2-symmetric
auxiliary 11 offers an enhanced stereoselectivity of both the
Ca-alkylation
step as well as for the iodolactonisation.
Several
groups developed polymer-bound oxazolidinones for asymmetric acyl group-based
chemistry, including aldol reactions, conjugate addition and enolate alkylations
as well as Diels-Alder cycloadditions. The first example of such an
immobilized Evans auxiliary was published by Allin and Shuttleworth and
applied in the synthesis of an a-alkylated
carboxylic acid (7). Oxazolidinone
12,
derived from (S)-serine, was treated with potassium hydride and
the resulting alkoxide was attached to Merrifield resin, followed by removal
of the BOC protecting group to furnish the polymer-bound auxiliary
13
(Scheme 3).
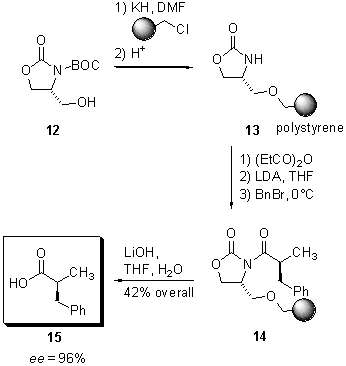
Scheme
3 Asymmetric synthesis of a-alkylated
propionic acid
Treatment
of polymer-bound oxazolidinone 13 with propionic anhydride under
basic conditions and subsequent a-alkylation
with LDA and benzyl bromide at 0°C gave rise to 14, from which a-benzylated
propionic acid 15 was released by saponification with lithium hydroxide.
The product was obtained in 42% overall yield and excellent enantioselectivity
(ee = 96%). The recycling of the auxiliary was not demonstrated.
It should be noted at this point, that subsequent work demonstrated some
difficulties to reproduce these results (8).
At
the same time, Burgess and Lim (9)
examined the stereochemical outcome of the same alkylation with a similar
oxazolidinone derived from (S)-tyrosine. For the immobilization
of the auxiliary, N-acylated oxazolidinone 16 was coupled
under Mitsunobu conditions to Wang and TentaGel resin or by nucleophilic
substitution to Merrifield resin (Scheme 4).

Scheme
4 Alkylation of oxazolidinone 17 immobilized on different solid
supports.
After
deprotonation with LDA and alkylation with benzyl bromide at 0°C, alcohol
18
was obtained by reductive cleavage from the solid support with lithium
borohydride in different yields and enantiomeric excesses, depending on
the nature of the solid support. While Merrifield resin, in contrast to
the results published by Allin and Shuttleworth, showed poor stereoselectivity
(ee up to 56%), the best results were obtained with the Wang resin
(ee up to 90%). Poorer yields (12-39%) and enantioselectivities
(ee = 71-88%) were observed for the alkylation with benzyloxymethyl
chloride (BOMCl) and aliphatic alkyl bromides.
The
first asymmetric aldol reaction on a solid support was performed by Purandare
and Natarajan (10). They used a
tyrosine based oxazolidinone 19 immobilized on Wang resin, prepared
by the protocol reported by Burgess et al., for the synthesis of a-substitutedb-hydroxy
ester 22 (Scheme 5).
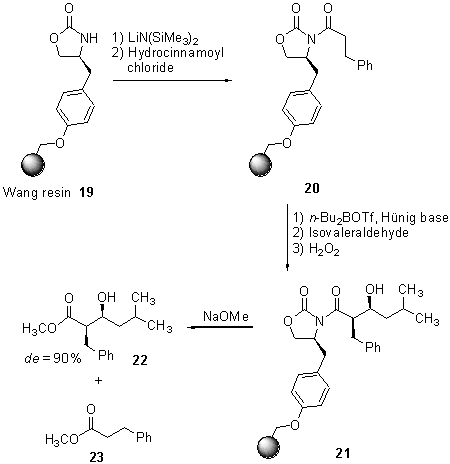
Scheme
5 Asymmetric aldol reaction on solid support
The
polymer-bound auxiliary 19 was acylated with hydrocinnamoyl chloride
and the following aldol reaction with isovaleraldehyd was investigated
under several conditions, i. e. changing of reaction temperature, number
of equivalents of boron reagent and Hünig base. Under optimized reaction
conditions, ester 22 was obtained along with hydrocinnamoyl ester
23,
derived from unreacted starting material, in a 90 : 10 ratio by detachment
from the polymeric support using sodium methoxide. HPLC and
1H-NMR
studies revealed that the product was predominantly a single syn
diastereomer (20:1), but either enantiomeric excess nor chemical yield
of the reaction sequence were given.
A
high reproducibility of the aldol reaction in solution to the solid support
in respect to stereoselectivity and chemical yield has been demonstrated
by Poon and Abell (11). Oxazolidinone
24,
immobilized on hydroxymethyl polystyrene resin, was acylated with propionyl
chloride after deprotonation with n-butyllithium and enolised
using n-dibutylboron triflate/triethylamine (Scheme 6).

Scheme
6 Solid-phase aldol and conjugate addition reactions
Subsequent
aldol reaction at -78°C
with benzaldehyde furnished a polymer-bound aldol adduct. After cleavage
from the polymeric support with lithium hydroxide in THF, b-hydroxy
acid 25 was obtained as a clean product (94% purity) in 63% overall
yield and with excellent diastereo- and enantioselectivity (de,
ee
> 98%). The authors also investigated the conjugate addition of the propionated
oxazolidinone 26 to acrylonitrile (Scheme 6). After enolisation
with TiCl3(Oi-Pr) and diisopropylamine, acrylonitrile
was added and the resulting product cleaved from the solid support using
lithium hydroxide in THF to furnish a-methylated
3-cyano butyric acid 27 in 52% overall yield, but with lower enantiomeric
excess (ee = 78%) as observed for the aldol reaction.
A
different approach for the asymmetric aldol reaction on solid support was
reported by Reggelin et al. (12).
A polymer-bound aldehyde, obtained from the oxidation of Wang resin, was
treated with the boron enolate of an acylated Evans oxazolidinone at -78°C.
After treatment with trimethyl aluminium and N,O-dimethylhydroxylamine,
the aldol adduct was cleaved from the solid support and isolated in 65%
overall yield and with a syn to anti ratio of 87:13. The
resulting polymer-bound Weinreb amide was then transformed into its corresponding
aldehyde and reused in the aldol reaction. This methodology could be extended
by the employment of a fluoride ion labile silyl linker on Merrifield resin
([i])
or a soluble polymeric support (14).
By this improvements, the cyclic reestablishment of the aldehyde functionality
allowed the iterative synthesis of di- and triketides in diastero- and
enantiomerically pure form.
Cycloadditions
are one of the most versatile reactions in organic synthesis on solid support
because of their ability to build up cyclic ring systems. Winkler and McCoull
(15) described the application
of the tyrosine-based oxazolidinone system 24 to the asymmetric
Diels-Alder cycloaddition. Oxazolidinone 24, prepared by attachment
of N-BOC-(S)-tyrosine methyl ester to hydroxymethyl Merrifield
resin under Mitsunobu conditions and subsequent formation of the oxazolidinone
moiety, underwent acylation with in situ generated trans-crotonic
anhydride to furnish resin 28 (Scheme 7).
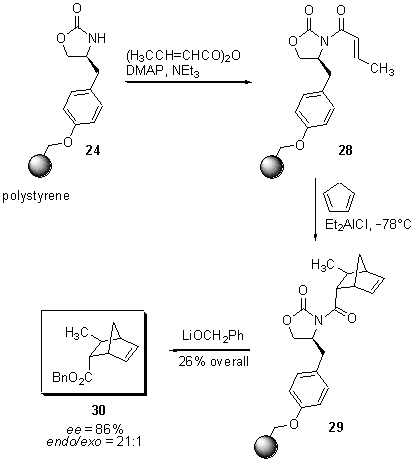
Scheme
7 Asymmetric Diels-Alder reaction on Merrifield resin
Diethyl
aluminium chloride catalyzed Diels-Alder reaction of dienophile 28
and cyclopentadiene at low temperature gave the bicycle 29, which
upon exposure to lithium benzylate released Diels-Alder adduct 30
from the solid support in 26% overall yield. The 21:1 endo/exo ratio
and the enantiomeric excess of ee = 86% compares favourably with
the selectivities obtained from the solution phase reaction (>20:1 endo/exo
ratio, ee = 88% ).
Recently,
another group reported the asymmetric 1,3-dipolar cycloaddition of resin
28
with mesitonitrile oxide and diphenylnitrone (16).
After reductive cleavage of the cycloaddition aducts from the solid support,
4,5-dihydro isoxazoles and isoxazolidines were obtained in good chemical
yields (up to 62%) and enantiomeric excesses (up to 89%). However, the
long reaction times (up to 40 days) and the formation of regioisomers are
a significant problem in this procedure.
A
combinatorial approach to a-substituted
glycine amides via Ugi four-component condensation (4-CC) on a solid support
was developed by Kunz et al. (17).
The chiral O-pivaloyl protected galactosylamine auxiliary 31
was prepared by the attachment of a galactosylazide via a heptanedioic
acid-based spacer to Wang resin, followed by reduction of the azide moiety
to generate the amino functionality. This strategy allows, after cleavage
of the ester linkage to the resin, the direct determination of the diastereomeric
excesses of the released amino acid derivative 33 (Scheme 8).
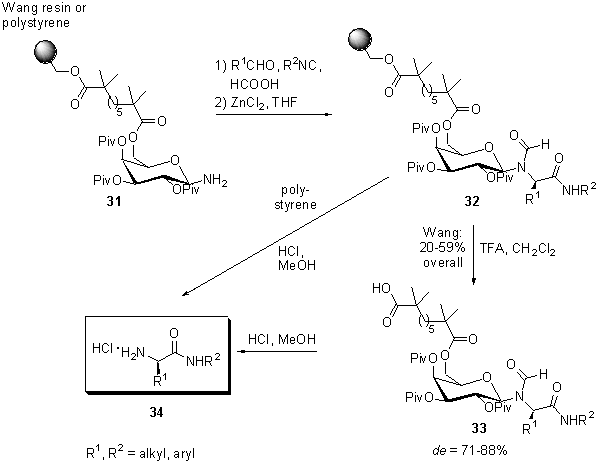
Scheme
8 Stereoselective Ugi four-component condensation
The
stereoselective Ugi four-component condensation, performed with five equivalents
of each aldehyde, isonitrile and formic acid under lewis acid conditions,
led to the formation of polymer-bound glycine amides 32. Acidic
detachment from the resin with trifluoracetic acid (TFA) in dichloro methane
furnished amino acid derivatives 33 in 20-59% overall yield and
diastereomeric excesses of de = 71-88%. After separation of the
diastereomers by preparative HPLC and cleavage of the N-glycoside
bond with hydrochloric acid in methanol, enantiomerically pure a-substituted
glycine amides 34 were obtained. The generality of the method was
demonstrated with a set of five different aldehydes and three isonitriles.
The employment of hydroxymethyl polystyrene resin instead of Wang resin
leads to a more acid stabile ester linkage, which allows the direct cleavage
of the glycine amides 34 from the solid support under the conditions
described above.
The
oxidative cleavage of polymer-bound sulfoximines to their corresponding
sulfones was reported by Hachtel and Gais (18)
(Scheme 9).
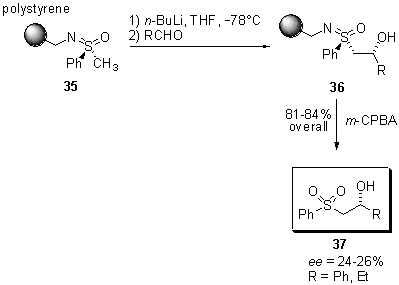
Scheme
9 Hydroxyalkylation of polymer-bound sulfoximines
Starting
from sulfoximine 35, readily available from Merrifield resin and
the potassium salt of (S)-S-methyl-S-phenylsulfoximine,
deprotonation in a-position
with n-butyllithium at -78°C
in THF and subsequent hydroxyalkylation with benzaldehyde or propanal led
to the formation of b-hydroxy
sulfoximines 36. Oxidative cleavage with m-chloroperbenzoic
acid proceeded smoothly to afford b-hydroxy
sulfones 37 in 81 and 84% overall yield, respectively. As already
expected from the results of the hydroxyalkylation of lithiosulfoximines
in solution phase, the asymmetric inductions are low and sulfones 37
were obtained with ee values of 26 and 24%.
Recently,
Enders et al. (19) reported the
development of two novel chiral hydrazine resins and their application
in the asymmetric synthesis of a-branched
primary amines. The enantiopure b-methoxyamino
auxiliaries, easily derived from amino acids trans-4-hydroxy-(S)-proline
and (R)-leucine, were attached to Merrifield resin and transformed
into their corresponding hydrazines 38 (SAMP-resin) and 39
(RAML-resin), representing immobilized analogues of the well-known SAMP
and (R)-methoxyleucinol auxiliaries (Scheme 10).
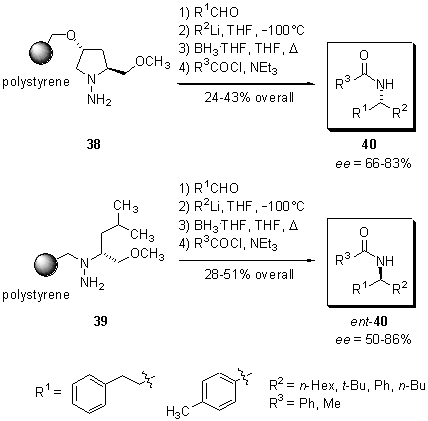
Scheme
10 Synthesis of acylated a-branched
primary amines.
Immobilisation
of various aldehydes, followed by nucleophilic 1,2-addition of aliphatic
and aromatic organolithium reagents at -100°C
in THF to the resulting hydrazones and subsequent reductive cleavage from
the solid support with borane-tetrahydrofuran complex, furnished a series
of a-branched
primary amines in 50-70% purity after extraction procedures. Further purification
was achieved by acylation of the amine moiety and chromatography of the
resulting amides 40, which were isolated in 24-51% overall yield
and with enantiomeric excesses of up to 86%. By choosing either the SAMP-resin
38
or the RAML-resin 39, the (R)- or (S)-amide
40
can be synthesized. In addition, this protocol shows great flexibility
regarding aliphatic and aromatic substrates and allows the synthesis of
the acylated a-branched
amines from commercially available substrates.
Summary and Conclusion
References
During
1-30 September 2001, all comments on this poster should be sent by e-mail
to [email protected]
with XXXX as the message subject of your e-mail. After the conference,
please send all the comments and reprints requests to the author(s).