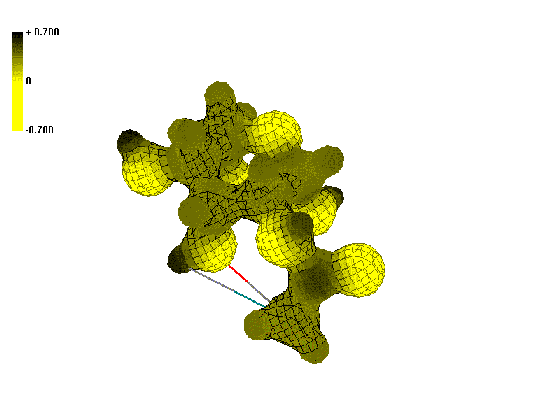
Figure 1. Isosurface of the electrostatic potential in the vicinity of N-acyl-2-AG (2)
Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0046]
Substitution Effects on Reactivity of
N-Acyl- 2-amino-2-desoxigliukopyranoses.
Quantum Chemical Study.
Aušra Vektarienė a, Arvydas Juodviršis a, Gytis Vektaris b
a
Institute of Biochemistry, Mokslininkų 12, 2600 Vilnius, Lithuania
Tel.: (370-2)729195, Fax: (370-2)729196, E-mail: [email protected]
b
Institute of Theoretical Physics and Astronomy, A. Goštauto 12, 2600 Vilnius, Lithuania
Tel.: (370-2)620953, Fax: (370-2)225361, E-mail: [email protected]
Received: 29
July 2000 / Uploaded: 31 July 2000
Abstract
Quantum mechanical calculations were carried out to study the molecular geometry and electronic structure of 2-amino-2-desoxigliukopyranose (AG) and N-acetyl-, N-ethanoyl-, series of N-phthalimidealkanoyl-AG . The total charge density, electrostatic potential, spatial distribution and positions of HOMO and LUMO of N-acyl-AGs with respect to their substitutes yield information on the reactivity of the molecules.
Introduction
2-Amino-2-desoxigliukopyranoses (AG) containing residues of N-acyl functional groups demonstrate unique chemical and biological behavior. Experimentally, structure of N- substitutes on 2-amido-AG have been shown to affect significantly reactivity of gliukopyranose skeleton. In same cases, their reactivity is significantly enhanced allowing for selective reaction which usually cause chemical or conformational rearrangements [1-3]. In other cases N-acyl-AG molecule becomes unreactive or destroys gliukopyranose ring which is synthetically disastrous [2]. It would be a significant benefit to synthetic carbohydrate chemist to have a theoretical protocol of quantum mechanical estimation of N-acyl-AG for a initial recognition of reactivity of compounds.
Results and discussion
Quantum mechanical calculations were carried out to
study the molecular geometry and electronic structure of 2-amino-AG 1 and N-acetyl 2, N-ethanoyl 3, series of N-phthalimidealkanoyl-AG 4-8
(Scheme 1) by HyperChem 5.0
package [4].
Full geometry optimizations were performed by the semi-empirical MNDO
method at the RHF level of theory. The binding energy and the energy of the highest
occupied molecular orbital (HOMO) and the lowest unoccupied
molecular orbital (LUMO), bond
lengths, angles and the charges of atoms finally were
calculated. The energy calculations of conformers 1-8
shows that a -anomers are lowest energy than b -anomers. This result is in agreement with C13 NMR
spectra which shows increasing of a -anomers at the expense of b -form [3]. Further calculations substituted and
unsubstituted AG 1-8 are based on a
-anomers conformations since these are lower in energy than b
-forms. The gliukopyranose skeleton for N-acyl-AG adopt the chair conformation with the
planar conformation of N-phthalimide rings.
The total charge density (Table 3),
electrostatic potential (Figure 1 and Figure 2),
spatial distribution and positions of HOMO (Figure 3, Figure
4, Table 1) and LUMO (Figure 5, Figure 6, Table 1) of N-acyl-AGs 1-8 with respect to their substitutes reflect information on
the reactivity of the molecules in an actual reactions with electrophiles or nucleophiles.
The representative plot of electron charge density, indicates a build up of positive
charge density on 1-C, 2-C and 3-C carbon atoms of N-acyl substituted pyranose skeleton 2-8 compared to unsubstituted 2-amino-AG 1. Figure 1 and Figure
2 presents the electrostatic potentials (ESP) of the N-alkanoyl and
N-phthalimidealkanoyl substituted AG.
Figure 1. Isosurface of the electrostatic potential in the vicinity of N-acyl-2-AG (2)
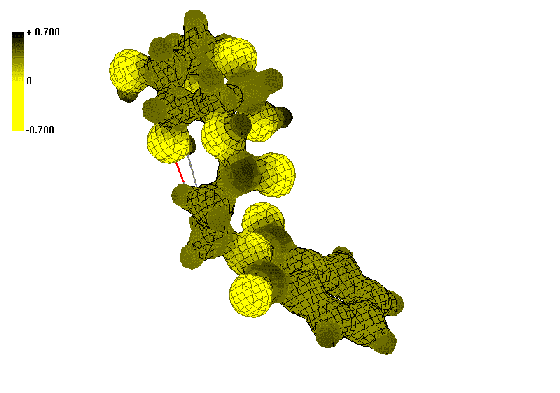
Figure 2. Isosurface of the electrostatic potential in the spatial vicinity of N-acyl-2-AG
(4)
Dark (black) colors indicate positive ESP regions and light (yellow)
colors indicate negative ESP regions. Comparison of ESP of the N-alkanoyl-AG 2, 3 (Figure 1) with the
N-phthalimidealkanoyl-AG 4-8 (Figure 2)
shows that for compounds 2, 3 increased positive charge
regions located on carbon atom of amide bond, while derivatives 4-8
gets more positive ESP regions on carbon atoms of CO-N-CO fragment of phthalimide ring.
Substitution on gliukopyranose skeleton of
N-acyl-AG 1-8 affects HOMO and LUMO energies (Table 1). All N-acyl substituents for 2-8
leads to decrease HOMO-LUMO gaps compared to the unsubstituted 2-amino-AG 1. Energy gap
reduction is caused by a strong decrease of LUMO energies while HOMO energies increase
slightly. The N-phthalimidealkanoyl substitution for 4-8
leads to smaller band gap than N-acetyl, N-ethanoyl substitution for 2 and 3 respectively, since
the LUMO energy for 4-8 is lowered more than for
compounds with N-acetyl, N-ethanoyl moieties 2, 3. The
strongest reactivity effect was predicted for the N-phthalimidealkanoyl substitution.
This result reflects the experimental data [3]. One can
therefore identify for N-acyl-AGs 2-8 the low lying LUMO
as a site will be most likely involved in reactions with nucleophile [5].
The spatial distribution of LUMO in N-acetyl, N-ethanoyl substituted AG
2, 3 are concentrated around C atom of amide that reflect
electron density are small. This site of molecule therefore most likely participate in
reactions with nucleophiles moieties. In the case of compounds 4-8
substituted with series of N-alkanoylphthalimide LUMO are located at the site of the
phthalimide ring on CO atoms. It shows that reaction with nucleophile most likely occur
close to CO on ring with subsequent cleavage of CO-N-CO fragment.
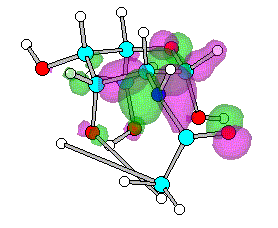
Figure 3. Optimized geometry and spatial distribution of HOMO for N-acyl-AG (2)
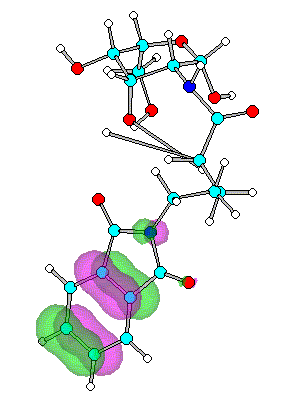
Figure 4. Optimized geometry and spatial distribution of HOMO for N-acyl-AG (6)
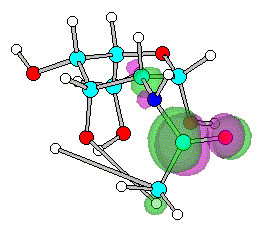
Figure 5. Optimized geometry and spatial distribution of LUMO for N-acyl-AG (2)
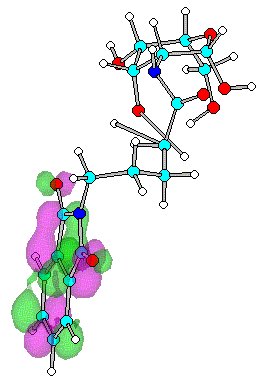
Figure 6. Optimized geometry and spatial distribution of LUMO for N-acyl-AG (6)
The molecular orbital analysis of electronic structure
of the N-acyl-AGs 2-8 and 2-amino-AG 1 correlated with experimental results [1-3].
N-acyl-AG are involved in reactions with nucleophiles in most cases causing decomposition
of amide bond. N-alkanoyl-AGs are reactive in rigid conditions which sometimes cause
destruction of gliukopyranose skeleton. AG substituted with N-phthalimide moieties
demonstrate selective reactivity in light condition. They can acts with hydrazine leading
to rearrangement on CO-N-CO fragment of phthalimide skeleton with the formation of
ammonium salts bearing phthaloylhydrazide while amide on gliukopyranose skeleton remains
unreactive [3].
Conclusions
Results of quantum chemical calculations reflect
information on the reactivity of N-acyl-2-amino-2- desoxigliukopyranoses.
Substitution on gliukopyranose skeleton affects HOMO and LUMO energies.
The N-phthalimidealkanoyl substitution leads to smaller HOMO-LUMO
energy gap than in case of the N-alkanoyl moieties.
Energy gap reduction caused by the strong decrease of LUMO energies. The strongest
reactivity effect with nucleophiles was predicted for the 2-amino-2-desoxigliukopyranoses
bearing N-phthalimidealkanoyl substitutes.
The spatial distribution of LUMO in the alkanoyl substituted
2-amino-2-desoxigliukopyranoses located on CO fragment of amide. In the case of
N-alkanoylphthalimide LUMO are located on CO atoms at the site of phthalimide ring. This
explains why different sites of N-acyl-2-amino-2- desoxigliukopyranoses will be most
likely involved in reactions with nucleophiles.
Acknowledgment
The authors thanks Dr. A. Stončius, Department of Organic Chemistry, Vilnius University for providing access to HyperChem 5.0.
References
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0046 as the message subject of your e-mail.