[B0002]
Mark Meisenbach, Thomas Allmendinger, Ching-Pong Mak
Novartis Pharma AG, Chemical & Analytical Development, CH-4002 Basel,
Switzerland.
[email protected]
Received: 27 July 2001 / Uploaded 7 August 2001
Introduction
Research in pharmaceutical industry and academia concentrate on combinatorial chemistry for lead finding and lead optimization. One of the practical approaches is the solid phase supported parallel synthesis to generate libraries for high-throughput screening. The number of hits may increase dramatically and the compounds as potential drugs are entering development. Process chemists are faced with the problem of rapid delivering the first amounts of drug substance necessary for early preclinical and clinical studies. Since the synthesis originated by a solid phase approach, no solution phase procedures for these compounds are available.
To tackle with this problem we were interested in gaining experience to scale up the solid phase syntheses directly. Time, an important factor in drug development, is saved by omitting the search for alternatives and by running solid phase multi step syntheses without isolation and purification of intermediates. Starting from a, b-unsaturated ketones 1 a library of different heterocycles was prepared in research [1].
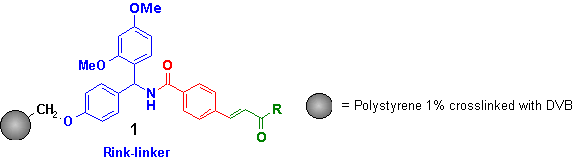
As an example for scale-up directly on the solid support the synthesis of 4-(2-amino-6-phenylpyrimidin-4-yl)benzamide 10 was chosen to study the following items:
(1) Scale-up phenomena of different solid phase reactions and on-bead analytics
(2) Effect of loading (an equivalent to concentration in solution phase chemistry)
(3) Selection of appropriate equipment
Results and Discussion
(1) Scale up phenomena and on-bead analytics
Due to the fact, that the Rink resin (attached via
an ether bond to the Merrifield resin as depicted in
1) is not available on large scale and extremely expensive, we used the
so called Fmoc Rink linker
4-{(R,S)-1-[1-(9H-fluoren-9-yl)methoxycarbonylamino]-(2,4-dimethoxybenzyl)}-phenoxyacetic
acid (3), well established in peptide synthesis and
therefore readily available and fairly cheap. 3 was coupled to aminomethyl polystyrene 1.54 mmol/g (2a,
AMPS), in a low excess (1.5 equiv.) using standard amid
forming conditions (see scheme). The degree of loading was determined by
UV [2], after the Kaisertest was negative. The
Fmoc group was cleaved with 20% diethylamine in DMF
and subsequently carboxybenzaldehyde (5) was coupled (diisopropyl
carbodiimide, HOBt). Using prolonged reaction times, the excess of building block 5 could be reduced by a factor
of 10 compared to the original protocol. The structure of the resulting
solid phase supported aldehyde 6 was confirmed by IR spectroscopy showing
a strong C=O band at 1710 cm-1 and the characteristic C-H Band at
2730 cm-1.

For the following Claisen-Schmidt reaction to chalcone (8) the research synthesis used a 20 fold excess of both LiOH*H2O and acetophenone (7) in dimethoxyethane on a 100 µmol scale (reaction time 16 h). Employing these conditions on 35 mmol scale, no conversion could be obtained after 22 h, as shown by IR spectroscopy. After sample cleavage with 20% TFA in dichloromethane only formylbenzamide (11) could be detected by HPLC.
Suggesting a low solubility of LiOH in DME under dry/aprotic conditions, we added some EtOH which initiated a fast reaction[3]. Samples cleavage from the resin showed the desired chalconamide, but also 20% Michael adduct 12, confirmed by LC-MS.
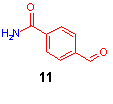
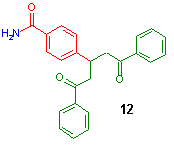
A short reaction screening provided considerable improvements: the reduction of the excess of acetophenone (7) and LiOH to 1.5 and 0.5 equivalents respectively, added to aldehyde resin 6a in a solvent mixture of THF and methanol gave complete conversion within one hour, with only 5% of Michael aduct as byproduct. For this Claisen-Schmidt reaction a Raman spectroscopic method was established to monitor the conversion online[4].
The final reaction steps could be scaled up without any problems: Chalcone 8a and guanidine (liberated from its hydrochloride with sodium ethanolate) are forming pyrimidine 9a by heating in dimethylacetamide (DMA) while bubbling air through the mixture for aromatization. After 16 h and complete conversion, the product was cleaved from the support (20% trifluoro acetic acid in DCM). The filtrate was evaporated and the residue upon recrystallization from ethanol/water gave the desired pure 4-(2-amino-6-phenyl-pyrimidin-4-yl)-benzamide 10 as the trifluoro acetate salt in 56% yield based on supported formylbenzoic acid.
(2) Effect of different loadings
As the solid support carried throughout the synthesis is finally ending up as waste, the ratio of solid support to product formed, can be diminished by increasing the number of attachment sites on the resin[5]. For the pyrimidine synthesis described, we looked for the effect of this loading quantified as mmol of functionality per g of resin. This was also a reason to switch from the ether-bonded Rink-linker to the acetic acid derivative to vary the loading of the resin easily by using aminomethylated polystyrenes (AMPS, 2) with different functionalization degrees. AMPS is commercially available with loading up to 2.9 mmol/g 2b (Novabiochem, Switzerland). A higher loaded resin was prepared by adapting and optimising the procedure of Adams[5]. Thus polystyrene cross-linked with 1% divinylbenzene is reacted with N-chloromethyl phthalimide in dichloromethane using iron (III) chloride as the Friedel-Crafts catalyst. By running the reaction at room temperature a colourless resin is obtained. The phthaloyl protection group was removed by means of aminolyses using methylamine in water/THF avoiding the common but toxic hydrazine.
The desired loading can be obtained by adjusting the stoichiometry ratio of PS/chloromethyl phthalimide/FeCl3. We synthezised a AMPS functioalized up to 4.48 mmol/g (2c), which means that 2 out of 3 phenylrings are aminomethylated, which has been not described so far.
To study the effect of loading we used 3 different resins
(1.54, 2.86 and 4.48 mmol/g) and repeated the synthesis of 10. The higher
loaded resins (2.86 and 4.48 mmol/g) didn't need any adjustments of conditions
found for the less loaded example already described. The quality of the product
was comparable and - as expected the ratio of product to support was
increased, as shown in the table.
Solid phase synthesis of pyrimidine 10
starting with resins of different loading
|
|
|
|
|
|
|
|
|
|
|
92% |
|
(>98%) |
|
|
|
|
66% |
|
(>97%) |
|
|
|
|
82% |
|
(> 98%) |
The results shown above indicate that solid phase supported synthesis already
optimized in research lab for the production of libraries on small scale can be
scaled up quite easily. In principle, reactors for scale-up of solid phase
peptide synthesis can be used. As peptide coupling and deprotection reactions
are highly optimized to run at room temperature the used suction filter
equipment does not have heating or cooling jacket. We therefore designed
reactors with temperature adjustments by incorporating sintered filter plates
into multiple necked double wall reactors with volumes up to 4 l. The outer
compartment can be connected to a heating/cooling circuit (thermostate).
Conclusion
References
1. E. Felder, A. Marzinzik, J. Org. Chem. 1998, 63, 723.
2. C. D. Chang, M. Waki, M. Ahmad, J.
Meienhofer, E. O. Lundell, J. D. Haug, Int. J. Pep. Prot.
Res.,1980, 15, 59.
3. C. Chiu, Z. Tang, J. W.
Ellingboe, J. Comb. Chem. 1999, 1, 73.
4. R. M. Dyson,
A. G. Helg, M. Meisenbach, T. Allmendinger, 11th International
Symposim on Pharmaceutical and Biomedical Analysis, Basle, May 14-18 2000, P
007.
5. S. P. Raillard, G. Ji, A .D. Mann, T. A. Baer, Org. Proc. Res.
Dev. 1999, 3, 177.
6. J. H. Adams, R. M. Cook, D. Hudson,
V. Jammalamadaka, M. H. Lyttle, M. F. Songster, J. Org. Chem.
1998, 63, 3706.