
Microwave Catalyzed Reaction of H-Dimethylphosphonate with N-(2, 3-Epoxypropyl)phthalimide
D. K. Rohrbaugh1, S. Munavalli2*, G. W. Wagner1, F. R. Longo2 and H. D. Durst1
1U. S. Army Edgewood
Chemical Biological Center, APG, MD 21010 (USA) and 2Geo-Centers,
Inc., P. O. Box 68, Gunpowder Branch, APG, MD 21010 (USA)
Phone:
410-436-2819; Fax: 410-436-3764; [email protected]
Received: 15 August 2001 / Uploaded 22 August 2001
Abstract
Microwave catalyzed reaction of N-(2, 3-epoxypropyl)-phthalimide with H-dimethylphosphonate) for four minutes yielded via free radical processes: trimethylphosphonate, trimethylphosphate, N-(2-propenyl)phthalimide, 2-(N-phthalimido-propyl) dimethylphosphinates, hydrogen 3-(N-phthalimidopropyl) methylphosphine oxide, hydrogen 3-(N-phthalimidopropyl)methylphosphinate, 2-(N-phthalimido-propyl) dimethyl-phosphonate and 2-(N-phthalimidopropyl) dimethyl-phosphinate. The mechanism of formation of the various compounds along with their mass spectral fragmentation behavior is described in this communication.
Introduction
Oxiranes comprise an extremely versatile group of intermediates and as such have attracted considerable interest in organic synthesis.1 However, their use in the reactions with phosphorus compounds has found only a limited number of applications including their routine utilization in the Michaelis-Becker reaction to prepare phosphinates.2, 3 Tri-coordinated pentavalent phosphorus compounds or in situ generated intermediates have been found to react with oxiranes.2, 3 Thus, phosphorus azide reacted with propylene oxide to furnish cyclic oxazaphoranes as well as acyclic compounds.4 Also, the in situ formed highly reactive metaphosphates have been described to open the oxirane ring to yield isomeric 1, 3, 2-dioxaphospholane-2-oxide derivatives.5
The in situ generated electrical energy from microwaves has been used to thermally catalyze chemical reactions. This type of energy transformation depends on the molecular properties of the reacting chemicals.6 Since the advent of commercially available microwave cookers, the microwave thermal process is finding increasing and interesting applications in synthetic organic chemistry.7 The popularity of the microwave-induced chemistry appears to rest primarily on its dramatic reduction of the reaction time and the possibility of carrying out neat reactions in "dry media" (solid phase). In fact, the latter has significantly contributed to its enhanced usage.8 The use of dielectric solvents seems to facilitate the transfer of the in situ generated thermal energy to chemical reactants8b. We became interested in adopting microwave chemistry for two reasons, namely the possibility of micro-scale chemistry and elimination of the hazardous waste thus generated during the normal work-up and its consequent disposal problems.
In continuation of our interest in the chemistry of the oxirane cleavage reactions,9 the microwave catalyzed oxirane ring opening in the presence of hydrogen dimethylphosphonate has been examined and observed to lead to the formation of unusual products. This paper describes the probable mechanism of the formation of the novel products formed during the said reaction and their GC-MS characterization.
Results and Discussion
Microwave catalyzed reaction of N-(2, 3-epoxypropyl)phthalimide (1, Fig. 1) with hydrogen dimethylphosphonate (2) has been examined and found to furnish ten compounds excluding the starting materials. Hydrogen dimethylphosphonate (2) itself gives rise to two compounds, namely trimethylphosphonate (3) and trimethylphosphate (4). There is nothing unusual about this, for these compounds are usually formed during the oxidation and/ or free radical reactions with hydrogen dimethylphosphonate (2). The presence of the readily removable hydrogen at the phosphorus center of the H-phosphonates appears to be the genesis of its reactivity.10 Among other things, H-phosphonates are known to be involved in the addition to: (i) multiple bonds11a and (ii) carbonyl group11b and (iii) in trans-esterifications11c. Dealkylations have also been noted [11b]. Both P O and C O bond cleavages have been observed.11d Methyl radicals have been stated to react with trimethylphosphite, albeit sluggishly, to give trimethylphosphonate.12 What is unusual about the reaction described herein is the non-specific radical formation from H-dimethylphosphonate. Recently, H-phosphonates have attracted considerable attention and have found useful applications in phosphorylation reactions.13
Deoxygenation of organic peroxides with phosphites has been described.14 However, epoxides have also been said to remain unaffected in the presence of phosphites.15 It has also been stated that phosphites deoxygenate epoxides to furnish alkenes.16 Thus, there seems to be some contradiction as regards the reaction of epoxides with phosphites. Phosphorus ylides, particularly carbanion-stabilized ylides, have been reported to react readily with the epoxides.17
The microwave catalyzed reaction of N-(2, 3-epoxypropyl)phthalimide (1) with H-dimethylphosphonate (2) has been found to yield several unusual compounds. This paper presents the formation and mass spectral characterization of the compounds thus formed. The presence of N-(2-propenyl)phthalimide (5) in the reaction mixture can be attributed to deoxygenation of the expoxide, 1. That N-(2-propenyl)phthalimide (5) is a product of the reaction and not an impurity in the starting material was ascertained by the GC-MS of the starting material. The mass spectrum of N-(2-propenyl)phthalimide (5) shows several interesting features. Next to the base peak, which happens to be the most intense peak, is the second most intense ion at m/e=169. This corresponds to the tricyclic ion (12), the genesis of which is described in Scheme 1. Similar loss of H2O has been reported for N-substituted phthalyl derivatives in rationalizing the formation of the tricyclic intermediate (13) (Scheme 1).18 In general, four characteristic and diagnostic ions can be seen in the mass spectra of phthalimido derivatives. They are m/e= 133, 130, 104 and 102. The ion at m/e=130 has been cited as the parent ion for the remaining two ions at m/e=104 and 102; which represent 2-cyanobenzoyl and 2-cyanophenyl entities (Scheme 2).19
The next compounds to come off the GC column are two stereomers with the molecular weight of 281 and with the retention times of 13.86 min and 14.13 min respectively. They exhibit almost identical mass spectral fragmentation patterns. They have been characterized as 2-(1-N-phthalylpropyl)dimethlphosphinate (6A and 6B or vice versa). It is rather difficult to single out and assign the correct structure for each of the two stereoisomers. Their mass spectral breakdown (Table 1) nicely accommodates the structures assigned to them. They both lose the phosphoryl entity when bombarded with electrons in the mass spectrometer to give N-(2-propenyl)phthalimide (5), which as described earlier goes on to give the ion at m/e=169 as the most intense peak.
Two phosphine oxide structures (7A
and 7B) were considered for the compound with M+=251. The mass
spectral fragmentation behavior of this compound led us to select structure
7A for this component, whose base peak happens to be the most intense
ion. However, the mass spectrum of this compound shows the peak neither at
m/e=169 nor at m/e= 160. Instead of the cleavage of N-alkyl side-chain, the
initial fission of the C _ N bond is observed, which is
followed by the loss of the CHO moiety to give the ion at m/e=222. The presence
of an ion at m/e=117 corresponds to the
[NC3H4PH(O)CH3] entity. Also seen in the mass
spectrum are the ions at m/e= 130 and 104.
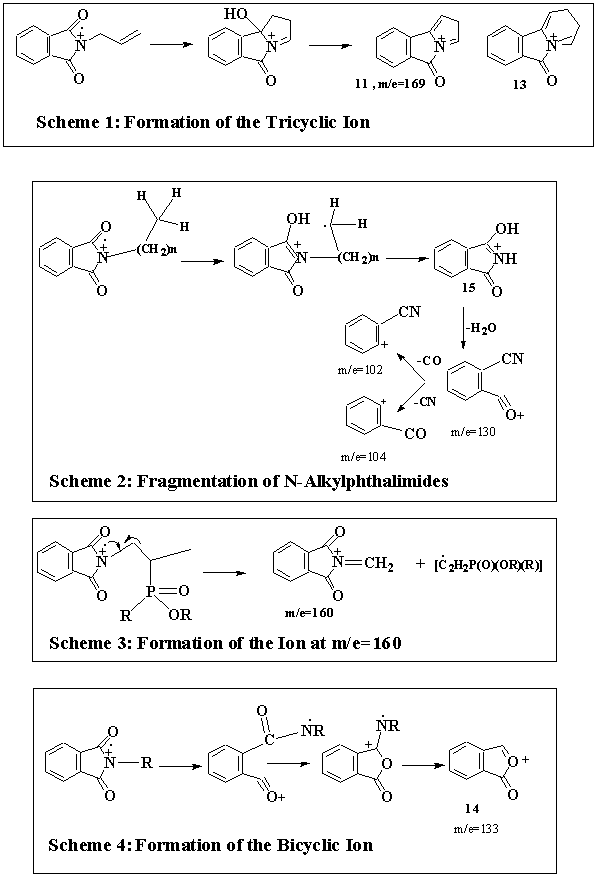
The most intense peak in the mass spectra of the remaining three compounds, namely 8, 9 and 10, happens to be the ion at m/e=160 (Scheme 3). This ion at m/e=160 (12, Fig. 1) has been previously proposed.18, 19b as arising from the cleavage of the N-alkyl side-chain accompanied by hydrogen transfer. These authors also propose the loss of the side chain with double hydrogen transfer to account for the formation of the intermediate ion (15) via the McLafferty rearrangement. This intermediate ion (15) has been suggested to serve as a precursor for ions at m/e=130, 104 and 102 (Scheme 2). The above cited three ions are seen in the mass spectra of all phthalimido-derivatives examined in this study. Some times, another ion at m/e=133 is seen in the mass spectra of the phthalimido-derivatives. A cyclic oxonium ion structure (14) has been assigned to this intermediate (Scheme 4).18, 19b
The component eluting at 16.02 min is a phosphinate with M+=267, for which structures 8A and 8B were considered as potential candidates. The ion at m/e=224 is due to the N-phosphinate entity, which results from the transfer of the phosphorus moiety to nitrogen of the phthalyl group. The phosphoryl moiety is lost from this compound to yield 5, which then goes on to form the most intense peak at m/e=160. Structure 8A has been assigned to this compound. Next comes the dimethylphosphonate with M+=297, for which two structures 9A and 9B were examined. The most intense peak of this component corresponds to ion, m/e=160 (Scheme 3). Also seen is a peak at m/e=137, [CH2CH2P(O)(OCH3)2]. Adding these two fragments, gives M+=297. This consideration led to 9A as the correct structure for this compound. Finally structure 10A was assigned to the last compound in preference to 10B. This choice was based on the presence of ions at m/e=185, 160 and 121. Adding of the last ions, namely m/e=160 and 121, gives 281, which is exactly the parent ion of this compound. Also seen is the fragment with m/e=185, which is M+ of N-(2,3-propenyl)phthalimide (5). These observations led directly to structure 10A as the correct structure of the last compound.
In summary, compounds 6 ~ 10
appear to have been formed from the addition of the various phosphoryl radical
intermediates to N-(2-propenyl)phthalimide (5), which itself has resulted
from deoxygenation of N-(2, 3-epoxypropyl)phthalimide (1). This inference
stands supported by the mass spectral characterization of to
N-(2-propenyl)phthalimide (5).

Experimental
Stoichiometric amounts of the respective reagents were mixed in glass vials or 5 ml ground joint round bottom flasks and tops, vigorously shaken on a vibro-mixter and heated in the microwave oven for a specified period. The reaction mixture was allowed to come to ambient temperature, the cooled product was first analyzed by gas chromatography and then subjected to GC-MS analysis.
All solvents were dry and freshly distilled prior to use. Mass spectra were obtained using a Finnigan TSQ-7000 GC/MS/MS equipped with a 30 m x 0.25 mm. i.d. DB-5 capillary column (J and W Scientific, Folsom, CA) or a Finnigan 5100 GC/MS equipped with a 15 m x 0.25 mm. i.d.Rtx-5 capillary column (Restek, Bellefonte, PA). The conditions on 5100 were: oven temperature 60-270° C at 10° C/min, injection temperature was 210°, interface temperature 230° C, electron energy 70 eV, emission current 500 mA and scan time 1 sec. The conditions on the TSQ-7000 were: oven temperature 60-270° C at 15° C/min, injection temperature 220°, interface temperature 250° C, source temperature 150° C, electron energy 70 eV (EI) or 200 eV (CI) and emission current 400 mA (EI) or 300 mA (CI) and scan time 0.7 sec. Data was obtained in both the electron ionization mode (range 45-450 da) and chemical ionization mode (mass range 60-450 da). Ultrahigh purity methane was used as the CI agent gas with a source pressure of 0.5 Torr (5100) or 4 Torr. Routine GC analyses were accomplished with a Hewlett-Packard 5890A gas chromatograph equipped with a J and W Scientific 30 m x 0.53 mm i.d. DB-5 column (J and W Scientific, Folsom, CA). The NMR spectra (1H and 13C) were recorded in CDCl3 with TMS as the internal standard on a Varian VXR-400S spectrometer at 100 MHz and 376 MHz respectively.
Microwave Catalyzed Reaction of N-(2, 3-epoxypropyl)phthalimide (1) with H-Dimethylphosphite (2): Stoichiometric amounts of H-dimethylphosphite (2, 0.110 g., 1 mmol) and N-(2, 3-epoxypropyl)phthalimide (1, 0.203 g., 1 mmol) were mixed in a glass vial or glass joint round bottom flask (5 ml), the mixture was shaken for a few minutes using the vibro-mixter and then heated in a table-top microwave oven for two minutes. The reaction mixture after cooling was analyzed by gas chromatography. Then, it was heated again for two minutes and re-analyzed. When no additional peaks showed up in the chromatogram, it was then subjected to GC-MS analysis. Thus the following compounds were characterized based on their mass spectral fragmentation behavior: (1) trimethylphosphonate (3), (2) trimethylphosphate (4), (3) N-(2-propenyl)phthalimide (5), (4 and 5) 2-(N-phthalimidopropyl) dimethyl-phosphinates (6A and 6B), (6) hydrogen 3-(N-phthalimidopropyl)methylphosphine oxide (7A), (7) hydrogen 3-(N-phthalimido-propyl)methyl phosphinate (8A), (8) 2-(N-phthalimidopropyl) dimethylphosphon-ate (9A) and (9) 2-(N-phthalimido-propyl) dimethylphosphinate (10A). The mass spectral data is given in Table 1.
References
1. (a) L. G. Lewis in "Comprehensive Heterocyclic Chemistry", vol. 7. , A. R. Katritzky, C. W. Rees, W. Lawoski (eds.), Pergamon Press, New York, 1984, p.100; (b) J. G. Buchanon, H. Z. Sable in "Selective Organic Transformations", vol. 2, B. S. Thyagarajan (ed), Wiley, New York, 1972, p. 1; (c) M. Bartok and K. C. Long in "The Chemistry of Ethers, Crown Ethers, Hydroxy Groups and their Sulfur Analogs", Part 1, Suppl., S. Patai (ed), Wiley, New York, 1980, p.609; (d) G. Smith, J. Chem. Soc., Chem. Comm. 629 (1984); (e) C. Bonini, R. DiFabio, G. Sotgiu, and S. Cavgnero, Tetrahedron 45, 2895 (1989); (f) A. S. Rao, S. K. Paknikar and J. G. Kirtane, Tetrahedron 39, 2323 (1983); (g) K. Maruko, M. Hasegawa, H. Yamamoto, K. Suzuki and G. Tsuchihashi, J. Am. Chem. Soc. 108, 3827 (1986); (h) K. Maruko, S. Nagahara, T. Ooi, and H. Yamamoto, Tetrahedron Lett. 30, 5607 (1989); (i) C. Bonini and G. Righi, Synthesis 225 (1994).
2. A. G. Rowley in "Organophosphorus Reagents in Organic Synthesis, J. I. G. Cadogan, Academic Press, NewYork (1979), p.306.
3. A Guide to Organophsophorus Chemistry, L. D. Quin, Wiley-Interscience, New York (2000).
4. G. Bertrand, J.-P. Majoral and A. Baceiredo, Tetrahedron Lett. 21, 5015 (1980).
5. R. Bodalski and L. D. Quin, J. Org. Chem. 56, 2666 (1991).
6. D. M. P. Mingos and D. R. Baghurst, J. Chem. Soc., Chem. Soc. Rev. 20, 1 (1991).
7. (a) R. J. Gedye, F. Smith, K. Westaway, H. Ali, L. Baldisera, L. Laberge and J. Rousell, Tetrahedron Lett. 27, 279 (1986); (b) R. J. Giguere, T. L. Bray, S. M. Duncan and G. Majetich, Tetrahedron Lett. 27, 4945 (1986); (c) A. Abramovitch, Org. Prep. Proc. Int. 23, 685 (1991); (d) S. Caddick, Tetrahedron 51, 10403 (1995); (e) P. de la Cruz, E. Diez-Barra, A. Loupy and F. Langa, Tetrahedron Lett. 37, 1113 (1996); (f) A. Dandia, H. Teneja, R. Gupta and S. Paul, Synth. Comm. 29, 2323 (1999); (g) B. K. Banik, M. S. Manhas, S. N. Newaz and A. K. Bose, Bioorg. Med. Chem. Lett. 3, 2363 (1993).
8. (a) P. Kumar and K. C. Gupta, Chem. Lett. 635 (1996); (b) S. Jolivet, S. A.-E. Ayoubi, D. Mathe, F. T. Boullet and J. Hamelin, J. Chem. Res(s), 300 (1996).
9. (a) S. Munavalli, D. I. Rossman, D. K. Rohrbaugh and H. D. Durst, National Meeting, American Chemical Society, Anheim (CA) 1995; S. Munavalli, D. K. Rohrbaugh, D. I. Rossman, L. R. McMahon and H. D. Durst, J. Organometal. Chem. 587, 160 (1999); (c) S. Munavalli, D. I. Rossman, D. K. Rohrbaugh and H. D. Durst (under preparation).
10. (a) P. J. Garegg, I. Smith, T. Redberg, J. Strowinski and R. Stromberg, Tetrahedron Lett. 27, 4051 (1986); (b) P. Westerduin, G. H. Veeneman, G. A. van der Marcel and J. H. van Boom, Tetrahedron Lett. 27, 6271 (1986).
11. (a) Methoden der Oragnischen Chemie, E. Muler (ed), Houben-Weyl, Georg Theime, Struttgart (1964) p.463; (b) M. S. Kharasch, R. A. Mosher and I. S. Bengelsdorf, J. Org. Chem. 25, 1000 (1960); (c) K. Troev and G. Borris, Phosphorus and Sulfur 29, 129 (1987); (d) W. Gerrard, W. J. Green and R. A. Nutkins, J. Chem. Soc. 4067 (1952).
12. (a) D. A. Bafus, E. J. Gallegos and R. W. Kiser, J. Phys. Chem. 70, 2614 (1966); (b) J.-J. L. Fu and W. G. Bentrude, J. Am. Chem. Soc. 94, 7710 (1972).
13. T. Wada, A. Mochizuki, Y. Sato and M. Sekine, Tetrahedron Lett. 39, 7123 (1998); (b) Y. Hayakawa in "Comprehensive Organic Synthesis", Vol. 5, B. M. Trost and I. Flemming (eds), Pergamon Press, New york (1991),p. 601; (c) J. Stawinski in "Handbook of Organophosphorus Chemistry", R. Engel (ed), Dekker Publishers, New York (1992), p.377; (c) L. D. Quin, "A Guide to Organophosphorus Chemistry", Wiley-Interscience, New york (2000).
14. (a) J. Krusic, W. Mahler and J. K. Kochi, J. Am. Chem. Soc. 94, 6033 (1972); (b) A. G. Davies, D. Griller and B. P. Roberts, J. Chem. Soc., Perkin Trans,. ll, 993 (1972); (c) K. J. Humphris and G. Scott, J. Chem. Soc., Perkin Trans,. ll, 831 (1973).
15. (a) G. O. Pierson and O. A. Runquist, J. Org. Chem. 34, 3654 (1969); (b) Y. Ito, M. Oda and Y. K. Kitahara, Tetrahedron Lett. 239 (1975); (c) C. H. Foster and G. A. Betchtold, J. Org. Chem. 40, 3743 (1975).
16. C. B. Scott, J. Org. Chem. 22, 1118 (1957).
17. (a) D. B. Denny, J. J. Vill and M. J. Borkin, J. Am. Chem. Soc. 84, 3944 (1964); (b) R. M. Gerkin and B. Rickborn, J. Am. Chem. Soc. 89, 5850 (1967); (c) W. E. McEwen, A. Blade-Font and C. A. Vander Werf, J. Am. Chem. Soc. 84, 677 (1962).
18. A. T. Aplin and J. H. Jones, J. Chem. Soc., Chem. Comm. 261 (1967).
19. (a) R. A. W. Johnstone, B. J. Millard and D. S. Millington, J. Chem. Soc., Chem. Comm. 600 (1967); (b) J. L. Cotter and R. A. Dine-Hart, J. Chem. Soc., Chem. Comm. 809 (1966).