Microwave Induced Reaction of H-Dimethylphosphonate with Styrene Oxide
S. Munavalli1*, D. K. Rohrbaugh2, G. W. Wagner2, F. R. Longo1 and H. D. Durst2
1Geo-Centers,
Inc., P. O. Box 68, Gunpowder Branch, APG, MD 21010 (USA) and 2U. S.
Army Edgewood Chemical Biological Center, APG, MD 21010 (USA)
Phone:410-436-2819; Fax:
410-436-3764; SXMUNAVA @SBCCOM.apgea.army.mil
Received: 15 August 2001 / Uploaded 22 August 2001
Keywords: styrene oxide, H-phosphonate, hydrophosporylation, free radical reaction
Introduction
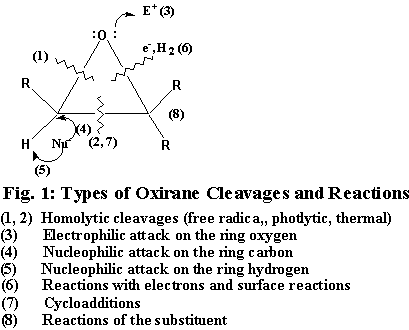
Oxiranes comprise an extremely versatile group of intermediates and as such have attracted considerable attention (1). Because of their ready availability and exceptional reactivity, the epoxides have found varied applications in synthetic organic chemistry. The oxirane ring can be opened under almost all conditions: electrophilic, nucleophilic, neutral, gas-phase, thermal and free radical conditions (Fig. 1) (1a). An excellent review on the preparation and synthetic applications of the oxiranes has appeared (1f). Recently we investigated the free radical cleavage of styrene oxide with trifluoromethylthiocopper and reported the formation of products arising from the C C and C O bond fission (2). However, their reaction with phosphorus compounds has found only a limited application including their routine use in the Michaelis-Becker reaction (3, 4). Tri-coordinated pentavalent phosphorus compounds or in situ generated intermediates have been found to react with oxiranes (3, 4). Thus, phosphorus azide reacted with propylene oxide to give cyclic oxazaphoranes as well as acyclic compounds (5). Also, highly reactive metaphosphate intermediates have been described to open the oxirane ring to yield isomeric 1, 3, 2-dioxaphospholane-2-oxide derivatives (6).
The in situ generated electrical energy from microwaves has been used to thermally catalyze chemical reactions. This type of energy transformation depends on the molecular properties of the reacting chemicals (7). Since the advent of commercially available microwave cookers, the microwave thermal process is finding increasing and interesting applications in synthetic organic chemistry (8). The popularity of the microwave-induced chemistry appears to rest primarily on its dramatic reduction of the reaction time and the possibility of carrying out reactions in "dry media" (solid phase). In fact, the latter appears to have significantly contributed to its enhanced usage (9). We became interested in adopting microwave chemistry for two reasons, namely the possibility of micro-scale chemistry and elimination of the hazardous waste generation during the routine procedure and work-up and its consequent disposal. In continuation of our interest in the chemistry of the oxirane cleavage reactions (10), the microwave catalyzed oxirane ring opening in the presence of hydrogen dimethylphosphonate has been examined and observed to lead to unusual products. This paper describes the probable mechanism of the formation of the novel compounds and their GC-MS characterization.
Results and Discussion
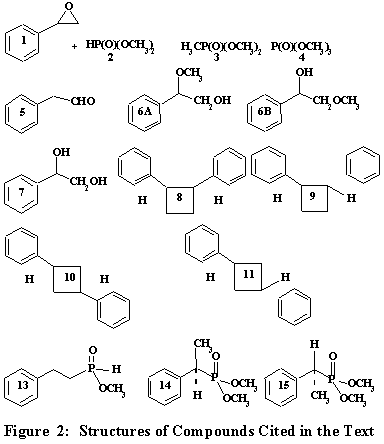
Recently, H-phosphonates have attracted considerable attention in phosphorylation reactions (11). Microwave induced reaction of styrene oxide (1) with hydrogen dimethylphosphonate (2) has been found to furnish ten compounds excluding the starting materials (Fig. 2). Hydrogen dimethylphosphonate (2) itself gives two compounds, namely trimethylphosphonate (3) and trimethylphosphate (4). There is nothing unusual about this, for they are usually formed during the oxidation and/ or free radical reaction of hydrogen dimethylphosphonate (2). The presence of the readily removable hydrogen at the phosphorus center of the H-phosphonates appears to be the genesis of its reactivity (12). Among other things, H-phosphonates are known to be involved in the addition to: (i) multiple bonds (13a) and (ii) carbonyl group (13b) and (iii) in trans-esterifications (13c). Dealkylations (13b). as well as P O and C O bond cleavages (13d) have been observed. Methyl radicals have been stated to react with trimethylphosphite, albeit sluggishly, to give trimethylphosphonate (14). What is interesting and unusual about the reaction described here is the non-specific radical formation from H-dimethylphosphonate (2).
 Deoxygenation of organic peroxides with phosphites
has been described (15). However, epoxides are said to remain unaffected in the
presence of phosphites (16). It has been stated that phosphites (17a) and
phosphines (17b) deoxygenate epoxides to give alkenes. Thus, there seems to be
some contradiction as regards the reaction of epoxides with phosphorus
compounds. Phosphorus stabilized carbanions are said to give various products on
reacting with oxiranes. Thus, the formation of alkenes, cyclopropanes and
ketones has been rationalized (18a). Significant formation of the ketones was
observed via hydrogen migration (18b). However, the treatment of styrene oxide
with benzylidene trimethylphosphorane yielded (2-phenylethyl)ketone as the minor
product and cis and trans 1,3-diphenylpropene as the major product (18b). With
methylenetriphenylphosphorane, styrene oxide gave a ketone and
triphenylphosphine. Strongly basic ylides have been reported to yield cyclic
ethers (18c). The reaction of styrene oxide with ethoxycarbonyl
triphenylphosphorane yields cyclopropanoids (18d-e).
Deoxygenation of organic peroxides with phosphites
has been described (15). However, epoxides are said to remain unaffected in the
presence of phosphites (16). It has been stated that phosphites (17a) and
phosphines (17b) deoxygenate epoxides to give alkenes. Thus, there seems to be
some contradiction as regards the reaction of epoxides with phosphorus
compounds. Phosphorus stabilized carbanions are said to give various products on
reacting with oxiranes. Thus, the formation of alkenes, cyclopropanes and
ketones has been rationalized (18a). Significant formation of the ketones was
observed via hydrogen migration (18b). However, the treatment of styrene oxide
with benzylidene trimethylphosphorane yielded (2-phenylethyl)ketone as the minor
product and cis and trans 1,3-diphenylpropene as the major product (18b). With
methylenetriphenylphosphorane, styrene oxide gave a ketone and
triphenylphosphine. Strongly basic ylides have been reported to yield cyclic
ethers (18c). The reaction of styrene oxide with ethoxycarbonyl
triphenylphosphorane yields cyclopropanoids (18d-e).
The compounds identified herein are formed via the free radical processes. Under the reaction conditions, styrene oxide (1) rearranges to give phenyacetaldehyde (5). There are precedents for this (1). There are two sites on styrene oxide (1) for the methoxy radicals to react. The attack on the unsubstituted a-carbon of styrene oxide (1) leads to compound 6B, while the attack on the b-carbon would result in compound 6A.

Since 1-methoxy-2-phenyl-2-ethanol (6B) has been characterized by its mass spectral fragmentation pattern, it seems the attack on the a-carbon is favored. Compound 6B loses the methyl moiety to give the oxy radical intermediate, which then abstracts hydrogen to form phenylethyleneglycol (7). The latter compound has been identified by its mass spectrum. The next compounds to come off of the g.c. column are a pair of isomeric diphenylcyclobutanes, namely cis and trans 1, 2-diphenylcyclobutanes (8 and 9) or cis and trans 1, 3-diphenylcyclobutanes (10 and 11). Styrene (12) which is formed from deoxygenation of styrene oxide (1) seems to serve as the in situ source for 8 and 9 or 10 and 11. There are precedents for the suggested dimerization of styrene (12) (19). The choice between the two pairs of isomers was made by comparison of the library spectra (20). Accordingly, these hydrocarbon isomers are considered as cis and trans 1, 3-diphenylcyclobutanes (10 and 11). 2-Phenylmethyl-phosphinate (13) arises from the reaction of the in situ generated styrene (12) with methylphosphinyl radical formed from 2, followed by hydrogen abstraction. Finally, the last compounds to elute off of the GC-MS column are a pair of stereomers, dimethyl (1-phenylethyl)phosphonates (14 and 15). These are formed from the reaction of dimethylphosphonyl radical (21) with styrene (12), followed by hydrogen abstraction. The formation and properties of the various phosphorus radicals have been described (22). Their mass spectral fragmentation patterns are similar. Fig. 3 attempts to describe the probable mechanism of the formation of the compounds described in the narrative.
Experimental
Stoichiometric amounts of the respective reagents were mixed in glass vials or 5 ml ground joint round bottom flasks and stoppered, vigorously shaken on a vibro-mixer and heated in the microwave oven for a specified period. The reaction mixture was allowed to come to ambient temperature, the cooled product was first analyzed by gas chromatography and then subjected to GC-MS analysis.
Mass spectra were obtained using a Finnigan TSQ-7000 GC/MS/MS equipped with a 30 m x 0.25 mm. i.d. DB-5 capillary column (J and W Scientific, Folsom, CA) or a Finnigan 5100 GC/MS equipped with a 15 m x 0.25 mm. i.d.Rtx-5 capillary column (Restek, Bellefonte, PA). The conditions on 5100 were: oven temperature 60-270° C at 10° C/min, injection temperature was 210°, interface temperature 230° C, electron energy 70 eV, emission current 500 mA and scan time 1 sec. The conditions on the TSQ-7000 were: oven temperature 60-270° C at 15° C/min, injection temperature 220°, interface temperature 250° C, source temperature 150°, electron energy 70 eV (EI) or 200 eV (CI) and emission current 400 mA (EI) or 300 mA (CI) and scan time 0.7 sec. Data was obtained in both the electron ionization mode (range 45-450 da) and chemical ionization mode (mass range 60-450 da). Ultrahigh purity methane was used as the CI agent gas with a source pressure of 0.5 Torr (5100) or 4 Torr (TSQ-7100). Routine GC analyses were accomplished with a Hewlett-Packard 5890A gas chromatograph equipped with a J and W Scientific 30 m x 0.53 mm i.d. DB-5 column (J and W Scientific, Folsom, CA). The NMR spectra (1H and 13C) were recorded in CDCl3 with TMS as the internal standard on a Varian VXR-400S spectrometer at 100 MHz and 376 MHz respectively.
Microwave Catalyzed Reaction of Styrene oxide (1) with H-Dimethylphosphonate (2): Stoichiometric amounts of styrene oxide (1, 0.22 g., 2 mmol) and H-dimethylphosphonate (2, 0.22 g., 2 mmol) were mixed in a glass vial or glass joint round bottom flask (5 ml), the mixture was shaken for a few minutes using the vibro-mixer and then heated in a table top microwave oven for two minutes. The reaction mixture after cooling to ambient temperature was analyzed by gas chromatography. Then, it was heated again for two minutes and reanalyzed. This process was repeated one more time to a total of six minutes of microwave heating. When no additional peaks appeared in the g. c. chromatogram, it was then subjected to GC-MS analysis. Thus, the following compounds were characterized based on their mass spectral fragmentation behavior: (1) dimethyl methylphosphonate (3), (2) trimethylphosphate (4), (3) phenylacetaldehyde (5), (4) 1-methoxy-2-phenylethanol (6B), (5) 1-phenylethleneglycol (7), (6) cis- and trans-1,3-diphenylcyclobutanes (10 -11), (7) hydrogen 1-(2-phenylethyl)methylphosphinate (13), (8) (1-phenylethyl)dimethylphosphonate (14) and (1-phenylethyl)dimethylphosphonate (15). Their retention times, percentages of the yields of the compounds and mass spectral fragmentation are described in Table 1.

References
1. (a) L. G. Lewis in "Comprehensive Heterocyclic Chemistry", vol.7. , A. R. Katritky, C. W. Rees, W. Lawoski (eds.), Pergamon Press, New York, (1984), p.100; (b) J. G. Buchanon, H. Z. Sable in "Selective Organic Transformations", vol. 2, B. S. Thyagarajan (ed), Wiley, New York, (1972), p. 1; (c) M. Bartok and K. C. Long in "The Chemistry of Ethers, Crown Ethers, Hydroxy Groups and their Sulfur Analogs", Part 1, Suppl., S. Patai (ed), Wiley, New York, (1980), p.609; (d) G. Smith, Synthesis 629, (1984); (e) C. Bonini, R. DiFabio, G. Sotgiu, and S. Cavgnero, Tetrahedron 45, 2895 (1989); (f) A. S. Rao, S. K. Paknikar and J. G. Kirtane, Tetrahedron 39, 2323 (1983); (g) K. Maruko, M. Hasegawa, H. Yamamoto, K. Suzuki and G. Tsuchihashi, J. Am. Chem. Soc. 108, 3827 (1986); (h) K. Maruko, S. Nagahara, T. Ooi, and H. Yamamoto, Tetrahedron Lett. 30, 5607 (1989); (i) C. Bonini and G. Righi, Synthesis 225 (1994).
2. S. Munavalli, D. K. Rohrbaugh, D. I. Rossman, L. R. McMahon and H. D. Durst, J. Organometal. Chem. 587, 160 (1999).
3. A. G. Rowley in "Organophosphorus Reagetns in Organic Synthesis, J. I. G. Cadogan, Academic Press, NewYork (1979), p.306.
4. A Guide to Organophsophorus Chemistry, L. D. Quin, Wiley-Interscience, New York (2000).
5. G. Bertrand, J.-P. Majoral and A. Baceiredo, Tetrahedron Lett. 21, 5015 (1980).
6. R. Bodalski and L. D. Quin, J. Org. Chem. 56, 2666(1991).
7. D. M. P. Mingos and D. R. Baghurst, J. Chem. Soc., Chem. Soc. Rev. 20, 1, (1991).
8. (a) R. J. Gedye, F. Smith, K. Westaway, H. Ali, L. Baldisera, L. Laberge and J. Rousell, Tetrahedron Lett. 27, 279(1986); (b) R. J. Giguere, T. L. Bray, S. M. Duncan and G. Majetich, Tetrahedron Lett. 27, 4945 (1986); (c) A. Abramovitch, Org. Prep. Proc. Int. 23, 685 (1991); (d) S. Caddick, Tetrahedron 51, 10403 (1995); (e) P. de la Cruz, E. Diez-Barra, A. Loupy and F. Langa, Tetrahedron Lett. 37, 1113 (1996); (f) A. Dandia, H. Teneja, R. Gupta and S. Paul, Synth. Comm. 29, 2323 (1999); (g) B. K. Banik, M. S. Manhas, S. N. Newaz and A. K. Bose, Bioorg. Med. Chem. Lett. 31, 2363 (1993).
9. (a) P. Kumar and K. C. Gupta, Chem. Lett. 635 (1996); (b) S. Jolivet, S. A.-E. Ayoubi, D. Mathe, F. T. Boullet and J. Hamelin, J. Chem. Res(S) 300 (1996).
10. (a) S. Munavalli, D. I. Rossman, D. K. Rohrbaugh and H. D. Durst, National Meeting, American Chemical Society, Anheim (CA) (1995); (b) S. Munavalli, D. I. Rossman, D. K. Rohrbaugh and H. D. Durst (under preparation); (c) S. Munavalli, D. K. Rohrbaugh, F. R. Longo, F. J. Berg and H. D. Durst, 213th National Meeting, American Chemical Society, San Diego (CA). (2001); (d) D. K. Rohrbaugh, S. Munavalli, G. W. Wagner and H. D. Durst (in press).
11. T. Wada, A. Mochizuki, Y. Sato and M. Sekine, Tetrahedron Lett. 39, 7123 (1998); (b) Y. Hayakawa in "Comprehensive Organic Synthesis", Vol. 5, B. M. Trost and I. Flemming (eds), Pergamon Press, New york (1991), p. 601; (c) J. Stawinski in "Handbook of Organophosphorus Chemistry", R. Engel (ed), Dekker Publishers, New York (1992), p.377; (c) L. D. Quin, "A Guide to Organophosphorus Chemistry", Wiley-Interscience, New York (2000).
12. (a) P. J. Garegg, I. Smith, T. Redberg, J. Strowinski and R. Stromberg, Tetrahedron Lett. 27, 4051 (1986); (b) P. Westerduin, G. H. Veeneman, G. A. van der Marcel and J. H. van Boom, Tetrahedron Lett. 27, 6271 (1986).
13. (a) Methoden der Oragnischen Chemie, E. Muler (ed), Houben-Weyl, Georg Theime, Struttgart (1964) p.463; (b) M. S. Kharasch, R. A. Mosher and I. S. Bengelsdorf, J. Org. Chem. 25, 1000 (1960); (c) K. Troev and G. Borris, Phosphorus Sulfur 29, 129 (1987); (d) W. Gerrard, W. J. Green and R. A. Nutkins, J. Chem. Soc. 4067 (1952).
14. (a) D. A. Bafus, E. J. Gallegos and R. W. Kiser, J. Phys. Chem. 70, 2614 (1966); (b) J.-J. L. Fu and W. G. Bentrude, J. Am. Chem. Soc. 94, 7710 (1972)
15. (a) J. Krusic, W. Mahler and J. K. Kochi, J. Am. Chem. Soc. 94, 6033 (1972); (b) A. G. Davies, D. Griller and B. P. Roberts, J. Chem. Soc., Perkin Trans,. ll, 993 (1972); (c) K. J. Humphris and G. Scott, J. Chem. Soc., Perkin Trans,. ll, 831 (1973),.
16. (a) G. O. Pierson and O. A. Runquist, J. Org. Chem. 34, 3654 (1969); (b) Y. Ito, M. Oda and Y. K. Kitahara, Tetrahedron Lett. 239 (1975); (c) C. H. Foster and G. A. Betchtold, J. Org. Chem. 40, 3743 (1975).
17. (a) C. B. Scott, J. Org. Chem. 22, 1118 (1957); (b) M. J. Borkin and D. B. Denney, Chem. and Ind. (London), 330 (1959).
18.(a) S. Trippett, J. Chem. Soc., Quart. Rev. 17, 406 (1963); (b) W. E. McEwen, A. Blade´-Font and C. A. Vanderwerf, J. Am. Chem. Soc. 84, 677 (1962); (c) R. Huisgen and J. Wulff, Ber. 102, 1841(1969); (d) W. J. Wadsworth, J. and W. D. Emmons, J. Am. Chem. Soc. 83, 6330 (1961); (e) R. M. Gerkin and B. Rickborn, J. Am. Chem. Soc. 89, 5850 (1967) and refs. cited therein.
19. (a) C. G. Overberger, M. Valentine and J.-P. Anselme, J. Am. Chem. Soc. 91, 687 (1969); (b) D. N. Harpp and C. Heitner, J. Org. Chem. 35, 3256 (1970); (c) T. Asanuma, M. Yamamoto and Y. Nishijima, J. Chem. Soc., Chem. Comm. 608 (1975); (d) T. Li, A. B. Padias and H. K. Hall, Jr., Macromolecules 23, 3899 (1990) and refs. cited therein.
20. Library of Mass Spectra, NIST, Gaithersburg, MD.
21. J.-J. Fu and W. G. Bentrude, J. Am. Chem. Soc. 94, 7710 (1972).
22. Free Radicals in Organic Chemistry, J. Fossey, D. Lefort and J. Sorba, Wiley and Sons (New York), (1995).