
[A004]

Centre for Molecular Architecture, Central Queensland University, Rockhampton, Queensland, 4702, Australia, e-mail: [email protected]
Introduction
The N-bridged [n]polynorbornanes are a new class of N-bridged norbornanes that have become available using cycloaddition protocols developed by our group.
[1] The basis for the synthesis is a dipolar cycloaddition reaction involving benzonorbornadienes as the dipolarophile and the 1,3-dipole formed by the ring-opening of a 5-azabicyclo[2.1.0]pentane. This reaction (the aza-ACE reaction [2]) requires that the aziridine be N-substituted thereby leading to the corresponding N-substituted polynorbornane (the trident). There are limitations on the type of substituent that can be introduced into the trident (eg the CBZ groups fail) and it has been necessary to find alternative groups that can be removed from the trident if the NH-bridged systems are to be accessed. This paper presents the results of our recent endeavours toward this goal of preparing NH-bridged tridents, especially those containing more than one NH-bridge.
The Synthesis of Tridents
We coined the 'dentane' name to describe the series of exo,exo-fused norbornanes in which the bridges have a syn-facial relationship (
Figure 1). The trident series is a subsection of the dentane family of polynorbornanes containing 3-fused norbornanes. [3] They have been so named because the three syn-facial bridges in the [3]polynorbornanes are structurally similar to the top section of a trident.

The
aza-ACE reaction [4,5]
The ACE and
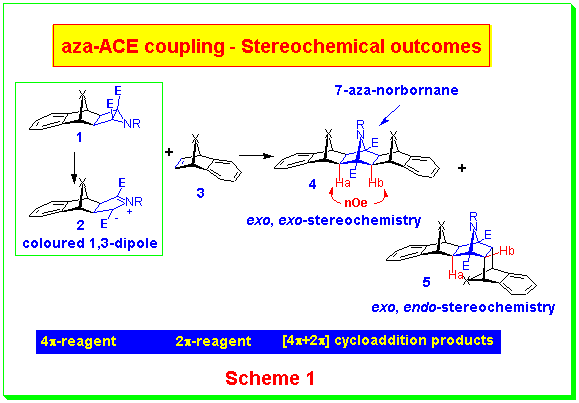
aza-ACE reactions are 1,3-dipolar cycloaddition reactions in which the 1,3-dipole is produced by ring opening of an ester-activated 5-oxabicyclo[2.1.0]pentane (ACE reaction) or an ester-activated 5-azabicyclo[2.1.0]pentane (the aza-ACE reaction). We have used the trident system as our testing ground for the aza-ACE reaction in order to assess coupling selectivities. The stereoselectivity of the aza-ACE coupling reaction has been evaluated using the aziridinocyclobutane 1 as the 4p-reagent and various benzonorbornadienes 3 as the 2p-reagents, following the early work on the ACE reaction. (Scheme 1).In the first stage of this work, we have been able to show that the aza-ACE coupling reaction is extremely tolerant of the bridging atom (or group) in the 7-position of the benzonorbornadiene and yields trident systems with various bridges combinations. The N-benzyl aziridine was used initially and this has since been extended to a variety of substituents on the aziridine nitrogen. The stereochemical outcomes are variable yielding both exo,exo-adducts (4) and exo,endo-adducts (5), the proportion of which depends on the N-substituent and the X-bridge of the dipolarophile.
The preparation of the cyclobutano-aziridine can be achieved in 3 steps starting from the appropriate benzonorbornadiene. This is illustrated in
Scheme 2 using benzonorbornadiene 6 (X=CH2) as the starting material. The initial step is common to both ACE and aza-ACE protocols and involves formation of the exo-fused cyclobutene 7 by ruthenium catalysed addition of dimethyl acetylenedicarboxylate (DMAD) (Mitsudo reaction). [6] Addition of the azide was achieved under high pressure conditions (RT, 3 days, 8-14 kbar). Conversion of the resultant triazoline 8 to the aziridine 9 is effected by photochemically-induced extrusion of dinitrogen (Rayonet reactor, l 300 nm, quartz) in benzene solution.

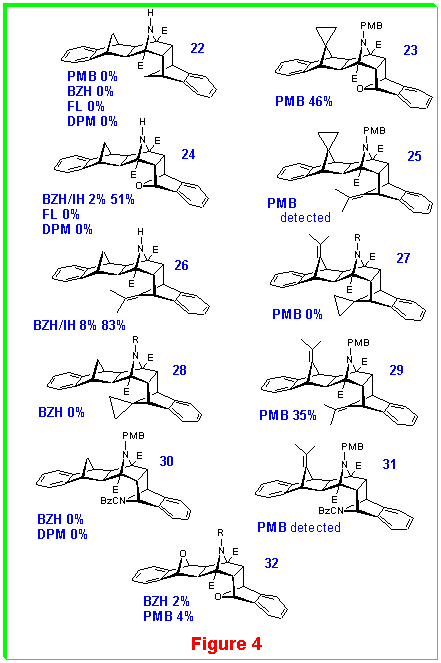
We summarize the results achieved to date on the synthesis of tridents where the central nitrogen is deprotected (see,
Figures 3 and 4). These molecules represent a novel class of heterocyclics with unknown physical and chemical properties and were prepared as model systems to study proximity effects, nitrogen inversion, protonation and metal complexation (Figure 2). [7,8,9]

In the benzyl aza-ACE reactions the trident systems were prepared using the 1,3-dipolar reaction. While over 25 N-benzyl tridents were prepared, deprotection at the central N-bridge could not be achieved under normal N-benzyl deprotection conditions. We attributed this to steric effects caused by the flanking bridges (sentinels) restricting reagent access to the central bridge nitrogen. In order to overcome this problem, we prepared the N-(methoxymethyl) (MOM) tridents, but attempts to remove the MOM groups initiated unexpected intramolecular transformations.
[10,11] In practice, the MOM method leads in some cases to nitrogen deprotection, but in general we have found that most initially formed MOM adducts are unstable and form secondary cyclisation products thereby rendering it unsuitable as a general synthetic route to NH tridents.We have now studied a series of aza-ACE reactions in which the N-substituent has been methoxymethyl (
MOM) [8,9] , benzhydryl [12] (BZH), p-methoxybenzyl (PMB) [11] , methoxycarbonyl (E) [10] , di-(p-methoxybenzhydryl) (DPM) [13] and 9-fluorenyl (FL) [11] . Different reaction conditions have been tested in order to remove the N-substituents such as treatment with acid (TFA), ionic hydrogenation (IH) [14], or catalytic hydrogenation (H2). The products of these reactions are displayed in Figures 3 and 4, together with their method of preparation and their isolated yields. Negative results are also included (attempted routes by various methods which failed). Thus far, there is no single N-protecting group which gives access to all NH-tridents, but by using a variety of different substituents it has been possible to build a substantial library of these interesting heterocycles.

Reaction mechanisms
MOM deprotection.
The de-(methoxymethylation) mechanism proposed elsewhere [9] and shown in Scheme 3 has some interesting features since it involves formation of the iminium ion 36. [15] Compression towards planar nitrogen by the flanking ‘sentinal’ groups of the tridents, would lower the transition state energy (ground state effect) for vicinal elimination of methoxide from 35 to form the iminium ion 36 and so this ion presents itself as a plausible intermediate in the formation of NH compound 38. Formation of product 38 involves a hydrolysis step via an alcohol intermediate 37 and preliminary evidence indicates that this probably occurred in the course of chromatographic work-up.

Benzylic deprotections.
A general acid catalysed mechanism for the deprotection of benzylic-type groups in 39 is proposed in Scheme 4. The various protecting groups that have been tried were chosen for their ability to stabilize the putative carbenium ion formed by the breaking of the critical N-bond to benzylic carbon. Of them all, the p-methoxybenzyl group was generally the most successful for the widest range of trident combinations but several of the others shown led to a variety of unexpected products which are not discussed here.

Conclusion.
The results outlined above for the new N- protected aza-ACE variants represent useful synthetic tools for preparation of tridents bearing a deprotected NH at the central bridge. The unknown physical and chemical properties of this novel class of heterocyclics are yet to be explored but, with their all syn- orientation of adjacent hetero atoms, offer exciting opportunities for protonation, alkylation or metal complexation studies.
Acknowledgements.
We thank the Australian Research Council (ARC) for funding and Mr. M. Hammond for technical assistance.
References.
[
1.] Warrener, R. N.; Butler, D. N.; Russell, R. A. Synlett 1998, 566.[
2.] ACE is an acronym for Acetylene Cyclobutene Epoxide[
3.] for more details see Warrener, R. N.; Butler, D. N.; Margetic, D.; Russell, R. A., ECHET98, Rzepa, H. S. and Kappe, O. (Eds), Imperial College Press, 1998, ISBN-981-02-3549-1 http://www.ch.ic.ac.uk/ectoc/echet98.[
4.] Butler, D. N.; Malpass, J. R.; Margetic, D.; Russell, R. A.; Sun, G.; Warrener, R. N. Synlett 1998, 588.[
5.] Sun, G. Ph.D. Thesis, Central Queensland University, 2001.[
6.] Mitsudo, T.; Kokuryo, K.; Shinsugi, T.; Nakagawa, Y.; Watanabe, Y. Takegami, Y. J. Org. Chem. 1979, 44, 4492.[
7.] Butler; D. N.; Hammond, M. L. A.; Johnston, M. R.; Sun, G.; Malpass, J. R.; Fawcett, J.; Warrener, R. N. Org. Lett. 2000, 2, 721.[
8.] Malpass, J. R.; Butler, D. N.; Johnston, M. R.; Hammond, M. L. A.; Warrener, R. N. Org. Lett. 2000, 2, 725.[
9.] Margetic, D.; Johnston, M. R.; Warrener, R. N.; Butler, D. N., Article 37, Electronic conference on synthetic organic chemistry (ECSOC-5), http://www.mdpi.org/ecsoc-5.htm, September 1-30, 2001, ISBN 3-906980-06-5 MDPI, Basel, Switzerland.[
10.] Hammond, M. L. A. unpublished results, CQU, 1999.[
11.] Warrener, R, N.; Margetic, D.; Butler, D. N.; Sun, G. Synlett 2001, 2, 202.[
12.] Preliminary results have been presented as a poster presentation at The second Brisbane biological and organic chemistry symposium (BBOCS-2), The University of Queensland, Brisbane, Australia, November, 30 2001.[
13.] Warrener, R. N.; Butler, D. N.; Margetic, D. unpublished results.[
14.] Kursanov, D. N.; Parnes, Z. N.; Kalinkin, M. I.; Loim, N. M. Ionic Hydrogenation and Related Reactions, Soviet scientific reviews supplement series chemistry, Vol. 1, 1985, OPA Ltd., Amsterdam.[
15.] Böhme, C.; Hacke, M. “Methyliminium Salts”, in Böhme, C.; Viehe, H. G. eds. “Iminium Salts in Organic Chemistry” Adv. Org. Chem. 1976, vol. 9, Wiley, N. Y. pg 156.