


7th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-7), http://www.mdpi.net/ecsoc-7, 1-30 November 2003
[C004]
Easy route to 4-benzyloxy-2-(a,a,a-2H3)-methyl phenol for
the synthesys of (8-2H3)-(all-rac)-d-tocopherol
Francesco Mazzini*, [a] Piero Salvadori, [a] Thomas Netscher[b]
[a] Dipartimento di Chimica e Chimica Industriale, Università di Pisa Via Risorgimento 35, 56126 Pisa, Italy E-mail: *[email protected]
[b] Research and Development, Roche Vitamins Ltd CH-4070 Basel, Switzerland E-mail: [email protected]
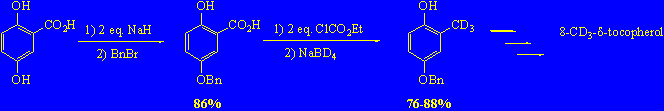
4d-tocopherol retrosynthesis : need of monoprotected hydroquinone derivative
44-benzyloxy-2-d3-methylphenol retrosynthesis
4Preparation by metalation reactions
4Preparation by multiple reductions reactions
44-benzyloxy-2-d3-methylphenol by two easy steps
Vitamin E (tocopherols and tocotrienols) is protective against many diseases (cancer, cardiovascular diseases[1], oxidation of low-density lipoproteins[2]), and is synthesized only in plants, making it a very important dietary nutrient for humans and animals[3]. Though a-tocopherol has the higher vitamin E activity, recent studies[4] showed unique g-tocopherol (gT) properties against prostate cancer and cardiovascular diseases, suggesting that the current vitamin E supplementation with primarily aT should be reconsidered. No such studies are reported, to our knowledge, on d-tocopherol, whose behaviour can be predicted similar by analogy reasons.
Therefore, in the last few years we have been focusing our attention on the preparation of deuterated tocopherols that greatly facilitate the understanding of their absorption and transport mechanisms. To improve the synthesis of d3-d-tocopherol[5], the preparation of 4-benzyloxy-2-d3-methyl phenol 1 represents the key point. Herein we report our studies that led to the tracing of an easy and very efficient route for its synthesis
d-tocopherol retrosynthesis : need of monoprotected hydroquinone derivative
Planning the preparation of d-tocopherol containing labels in the aromatic methyl group, in either racemic or stereoisomerically pure form, requires the protection of the phenolic group in position 4 to avoid the formation of 5- and 7-monomethyl regioisomers and possible double-alkylation products in the condensation step for the construction of chromanol ring. Protection is also needed in the synthesis of the halogenated derivatives (X=Br, I) that can be used in Heck coupling reactions, as recently successfully performed on iodo or bromo hydroquinones and allylic alcohol[6].
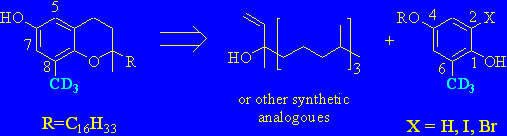
Considering the well-defined and effective synthesis already reported for the building of the aliphatic side chain[7], the key point of trideuteromethyl d-tocopherol total synthesis is the preparation of a suitable trideuterated hydroquinone derivative. Because of its stability under the condensation reaction and the mild conditions to be used for its subsequent removal, the benzyl group was believed to represent the best protective group for our purposes.
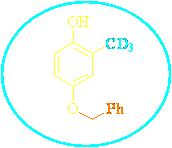
So our target was the synthesis of 4-benzyloxy-2-d3-methylphenol.
![]()
4-benzyloxy-2-d3-methylphenol retrosynthesis
On paper, there are different ways for preparation of 3. Deuterated methyl group could be introduced on substrate 5 or 2 by metalation reactions using CD3I as electrophile, having transformed the free phenolic group(s) with suitable DMG(s) (o-directing metalation group) group(s). Otherwise trideuteration could be achieved through a multi-steps reduction sequence starting from 7 or 4, easily synthesizable from commercially available 2,5-dihydroxybenzoic acid. Selective protection procedures have to be devised for the preparation of 3 from 6 and of 4 from 7.

![]()
Preparation by metalation reactions
Preparation of 3 by metalation of bis-DMG-hydroquinone was the first way investigated. 6 was obtained in high yield (up to 92%), but we were not able to separate the product from about 8-10% of the starting hydroquinone derivative. Moreover, selective protection of 6 resulted a quite difficult task. Direct metalation of 4-benxyloxy-1-DMG derivatives gave only 70% conversion in the best case (DMG=MOM). We obtained better results using bromine-lithium exchange at low temperature[8], though the preparation of 8 was tougher than expected. A thorough study was carried out employing n-BuLi and ter-BuLi as metalating agent, tuning the reaction conditions in terms of equivalents of metalating agent and electrophile, and time of electrophile addition. 1.4 equivalents of BuLi, 6 equivalents of CH3I added immediately after BuLi, 2 h represented the best result in our hands, with small amount of unreacted materials.

i) Br2, CHCl3, 25-88%; ii) 1.NaH, DMF 2. MOMCl, DMF; iii) BuLi, THF, -78°C ; CH3I, THF, -78°C.
![]()
Preparation by multiple reduction reactions
Successful examples for the benzylation of the least hindered phenolic group in 2,5-dihydroxybenzoic acid or its esters have been reported[9]. The aromatic ester moiety can be reduced to a methyl group through a multi-steps reduction sequence, with the corresponding benzylic alcohol 13 as an intermediate. Trials to perform further reduction of 13 without prior protecting the free phenolic group were unsuccessful.

i) MeOH, H2SO4, 96%; ii) K2CO3, BnBr, acetone, 69%; iii) LiAlH4/THF,87%. (LG º leaving group)
It is likely that the observed instability is due to the formation of o-quinone methides species producted after phenoxide ion generation in the reaction basic conditions, which are highly prone to polymerize. Therefore we tried to block these side reactions protecting the free phenolic group as MOM derivative 14. Surprisingly the crude product of the reaction of 14 with MOMCl was constituted by a mixture of the desired 15 and compound 16, whose formation is not of immediate rationalization. Anyway reduction by LiAlH4 of the mixture gave expected 17 in high yields.

i) 1. NaH (2 eq.), DMF 2. BnBr (1 eq.), DMF, 86%; ii) NaH, DMF 2. MOMCl, DMF; iii) LiAlH4, THF, 86% in respect of 14
After several trials, we succeeded to prepare the bromide 18, reacting 17 with MeSO2Cl, 2,6-lutidine and excess LiBr. Together we recoved the analogous chloride 19 (derived by competitive nucleophilic substitution by Cl- present in the reaction after the formation of sulfonate). Overall yield in respect to the starting alcohol was 85%.
18-19 mixture was thus reduced with LiAlH4 soon after its isolation (the mixture resulted relatively unstable stored at room temperature), affording 20 in very good yield (90%).
Standard deprotection procedure (MeOH, H+) afforded 21 in high yield.

i) MeSO2Cl, 2,6-lutidine, excess LiBr, THF; ii) LiAlH4, THF, 77% in respect to 17; iii) MeOH, H+, 90%.
![]()

4-benzyloxy-2-d3-methylphenol by two easy steps (Minami reduction)
As often happens in the field of research, we found a much more effective way to 1 after all the work done described above.
Minami and Kijima described an easy way for reduction of 3,4,5-trimethoxysalicylic acid to 2,3,4-trimethoxyphenol, converting it into a bis-ethoxycarbonyl derivative using ClCO2Et and then reducing it to the corresponding methylphenol with NaBH4[10] in a H2O/THF 1:1 mixture.
Preliminary experiments made following the original procedure gave only 50% yield, but the easiness of obtaining the desired product with a considerable saving of time and purification steps prompted us to continue investigating.
Hence we finally found that increasing the amount of NaBD4 up to 8 equivalents afforded the desired methylated product in 89 % yield. We verified that water is needed for the success of reaction, and that D2O has to be used to have maximum deuterium incorporation.
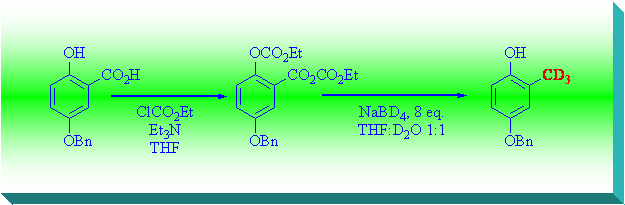
In the mechanism proposed by Mitchell[11], addition of hydride results in a benzyloxyanion that attacks the neighboring carbonate. Carbonate migration occurs and the resulting phenoxide undergoes a b-elimination thereby producing an o-quinone methide that is reduced by a hydride ion to afford the observed product.
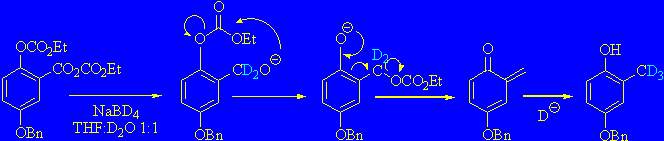
In our opinion, this procedure represents the simple conceivable way for the synthesis of 4-benzyloxy-2-(a,a,a-2H3)-methylphenol.
![]()
[1] K. F. Gey, P. Puska, Ann. N. Y. Acad. Sci. 1989, 570, 268-282.
[2] A. Kamal-Eldin, L. -Å. Appelqvist, Lipids 1996, 31, 671-701.
[3] G. Pongcraz, H. Weiser, D. Matzinger, Fat Sci. Technol. 1995, 97, 90-104.
[4] Q. Jiang, S. Christen, M.K. Shigenaga, B.N. Ames Am. J. Clin. Nutr. 2001, 714-722.M.G.Traber, L. Packer Am J Clin Nutr 1995, 62,1501S-9S.
[5] F. Mazzini, E. Alpi, Th. Netscher, P. Salvadori Eur. J. Org. Chem. 2003, 2003, 2840-2844.
[6] 6a J.Y. Goujon, F. Zammattio, B. Kirschleger Tetrahedron: Asymmetry 2000, 11, 2409-2420. 6b R.A. Outten, P. Bohrer, R.K. Müller, H. Schneider, A. Ruttimann, Th. Netscher Chemické Listy,18th Conference on Isoprenoids, 10-16 September 1999, Prachatice Czech Republic, P36.
[7] Th. Netscher Chimia 1996, 50, 563-567
[8] R. Kranich, K. Eis, O. Geis, S. Mühle, J.W. Bats and H. Schmalz Chem. Eur. J. 2000, 6, 2874-2894.
[9] U. Schmidt, H. Bokens, A. Lieberknecht, H. Griesser Tetrahedron Lett. 1981, 22, 4949-4952.
[10] N. Minami, S. Kijima Chem. Pharm. Bull. 1980, 28, 1648-1650.
[11] D. Mitchell, C.W. Doecke, L.A. Hay, Th.M. Koenig, D.D. Wirth Tetrahedron Lett. 1995, 36, 5335-5338.