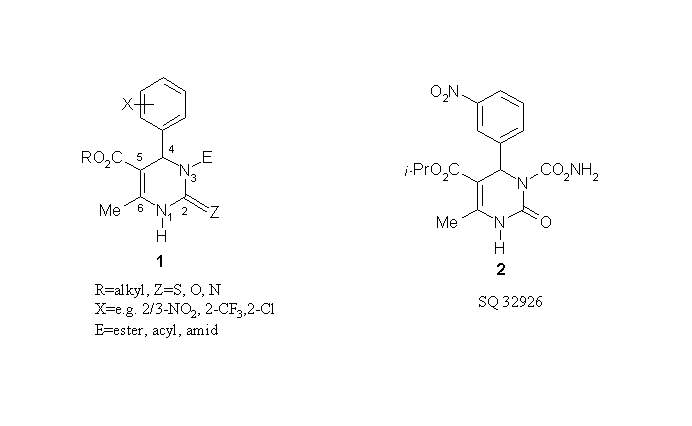
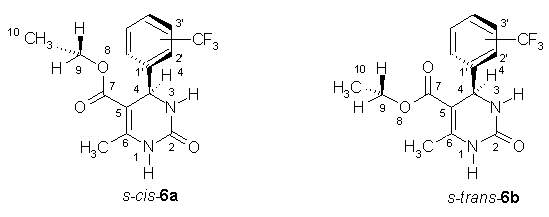
This study deals with theoretical prediction of vibrational spectra in
gaseous state and in CHCl3 solution of compounds 6a, 6b
at density functional B3LYP/6-31G* level. The Onsager reaction field
model4 has been employed to simulate the effect of solvent and in
addition, models with specific interactions were examined. The primary attention
was paid to stretching vibrations of both C=O bonds and N-H bonds. The results
are compared with experimental IR spectra. The conformers within both of series
6a and 6b arising from the orientation of CF3
substituent and CH3 group in ethoxy fragment were considered. The
nomenclature syn or anti distinguishes the relative orientation of
these groups with respect to the C4-H4 bond. The influence of conformation as
well as position of CF3 group in phenyl ring were investigated.
The calculated unscaled wavenumbers of CO stretching vibration in carbamate
fragment
ncar(CO), ester group
nes(CO) as well as NH stretching vibrations
ns(N1H) and
nas(N3H) are presented in
Table
1. Experimentally, in CHCl
3 as a solvent the CO stretching band
wih the lower wavenumber is assigned to CO of the carbamate fragment and the
higher one to the ester CO. The
nes(CO)
covers the range 1700 cm
-1 -1713 cm
-1 whereas
ncar(CO) is in interval 1645 cm
-1 1650
cm
-1. In contrast to this experimental assignment, B3LYP/6-31G*
calculations predict
ncar(CO)>
nes(CO) in the gaseous state. Possible reasons for
these discrepancies between theoretically and experimentally determined order of
CO vibrations are
- Effect of solvation (bulk solvent effects as approximated by the Onsager
reaction field model )
This model treats the electrostatic field effect of
solvent and assumes solute in a spherical cavity of radius a0.
- Specific solvent-solute interactions, which can not be simulated by
Onsager reaction field model. (e.g., hydrogen bonds between the CO group of
solute and the partially acidic hydrogen atom of CHCl3 molecules)
- Formation of dimers through intermolecular hydrogen bonds. Such
interactions could reduce the stretching vibration of carbamate CO group and
thus change the order of CO vibrations.
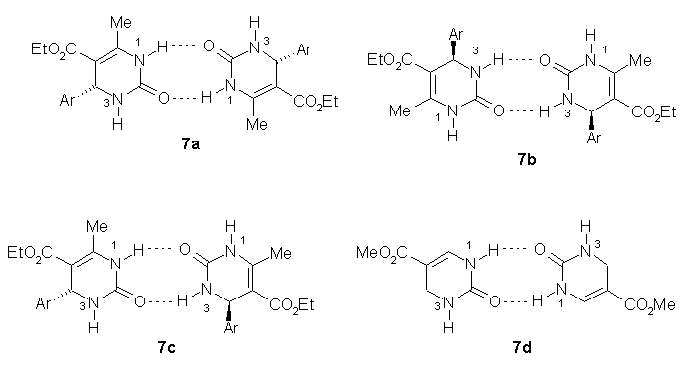
Three possible dimers
7a,
7b and
7c can be considered.
In IR spectra of analogous compounds (with unsubstituted phenyl ring) the
methylation of N1 atom leads to increase of
ncar(CO) from 1644,8 cm
-1 to 1676,4
cm
-1, which indicates that assumption of dimers in solution is
realistic and hydrogen of N1 is involved in the hydrogen bonding. This
encouraged us to use dimer
7a, but in order to reduced the computational
cost the calculations were performed for the dimer
7d (Figure 1) without
the aryl rings and methyl groups and with methoxy groups instead of ethoxy.

Figure 1 Structure of dimer 7d in gaseous state.
Moreover, measurements on the related compound
8 in different
solvents revealed the sequence of stretching vibrations
n(C2O)>
n(C7O) consistent with
that obtained by these calculations
5.
The presence of butyl substituent at nitrogen atom prevents the formation
of the dimers through hydrogen bonds. The hydrogen bonds between solute and
CHCl
3 group are also implausible since the wavenumber of C2=O
stretching mode in CHCl
3 solution (1704 cm
-1) is only 6
cm
-1 lower than in CCl
4 (1710 cm
-1) solution.
This shift can satisfactorily be attributed to the lower dielectric constant of
CCl
4. If C3 were replaced by nitrogen atom, the
n(C2O) could be expected at even higher value. This can be
deduced from the comparison of IR spectra of urea (
n(CO)=1734 cm
-1) with acetamide (
n(CO)=1728 cm
-1) measured in Ar-matrix
6
. The calculation on dimer
7d provides the order of CO stretching
vibrations in agreement with experiment. The theoretical
ncar(CO) is 1785 cm
-1 for symmetric
A
g mode and 1788 cm
-1 for asymmetric A
u mode
and the
nes(CO) is 1797 cm
-1 and
1802 cm
-1 for symmetric A
g and asymmetric A
u
mode, respectively. Besides treating the solvent as a contimuum, specific
solvent-solute interactions were taken into consideration too. At the
B3LYP/6-31G* level the associate of one representative solute with two molecules
of CHCl
3 (hydrogen bonds between hydrogen of CHCl
3 and the
oxygen of carbamate CO group (O
H)
carbamate and between the hydrogen
of the second CHCl
3 molecule and the oxygen of ethoxy group
(O
H)
ethoxy) was calculated. We supposed that the
(O
H)
carbamate could be responsible for the reduction of
ncar(CO) and (O
H)
ethoxy could enhance
the
nes(CO) to such an extent, that the
order of these vibrations would be consistent with the experimentally determined
one. The calculations on the complex in which all three oxygen atoms are forming
hydrogen bonds with three CHCl
3 molecules (Figure 2), were performed
at B3LYP/3-21G level on a model structure without aryl substituent. Although the
hydrogen bond (O
H)
carbamate was the strongest (CH stretching
vibration of CHCl
3 n(CH)=3048,6
cm
-1) whereas (O
H)
ethoxy was the weakest (
n(CH)=3158,9 cm
-1), it was not sufficient to alter
the sequence
ncar(CO)>
nes(CO).

Figure 2 Structure of associate with three molecules of
CHCl3 in gaseous state.
The calculations in CHCl
3 provide lower values for both the
ncar(CO) and
nes(CO) than those obtained for the gaseous state.
The
s-trans conformers possess lower
nes(CO) when compared to
s-cis. The
ns(N1H) and
nas(N3H) are systematically higher in series of
2-CF
3 positional isomers than in series of 3-CF
3
positional isomers. The stretching vibrations of N-H are coupled to each other.
The lower vibrations
nas(N3H) in asymmetric
mode are predominantly of N3-H character, while the higher vibrations
ns(N1H) in symmetric mode are mainly of N1-H
character. In
Table
1 the significant vibrational coupling is marked and is typical for
conformers with
syn directed CF
3 group in 2 position of
phenyl. In the case of such a coupling the values of
nas(N3H) and
ns(N1H) become closer to each other. The relative
energies with respect to the most stable conformer within both series are listed
in
Table
2 together with selected torsion angles. The effect of
syn or
anti orientation of CF
3 group on relative energies is more
pronounced in the set of conformers with CF
3 in 2 position, where
syn conformers are more stable. In set of conformers with CF
3
in 3 position the
s-cis or
s-trans orientation decides the
stability of conformers with
s-cis orientation being more favourable.
We have presented the result from density functional B3LYP/6-31G* study of
type s-cis-6a and s-trans-6b. The IR spectra are
predicted for gas and for solution in CHCl3 using the Onsager
reaction field model. The calculations on gaseous monomer as well as on monomer
in CHCl3 solution give the opposite order of CO stretching vibrations
than assigned experimentally. In order to resolve this discrepancy models for
specific solvent-solute interactions as well as formation of dimers were
applied. The associate formed between solute and solvents molecules through
hydrogen bonds left the order unaffected, whereas in the dimer the hydrogen
bonding altered the sequence consistent with experimental assignment. All
possible conformers were assumed and relative stability of them was determined.