8th International Electronic Conference on Synthetic Organic Chemistry. ECSOC-8. 1-30 November 2004. http://www.lugo.usc.es/~qoseijas/ECSOC-8/
| Quinaldine Derivatives Preparation and Their Antifungal Activity |
|
| Josef Jampilek1, Martin Dolezal1*, Jiri Kunes2, Vladimir Buchta3 |
1 Department of Pharmaceutical Chemistry and Drug Control, Charles University in Prague, Faculty of Pharmacy in Hradec Kralove, 500 05 Hradec Kralove, Czech Republic
e-mail: [email protected], tel.: +420-49-5067272, fax: +420-49-5512423
2 Department of Inorganic and Organic Chemistry, Charles University in Prague, Faculty of Pharmacy in Hradec Kralove, 500 05 Hradec Kralove, Czech Republic
3 Department of Biological and Medical Sciences, Charles University in Prague, Faculty of Pharmacy in Hradec Kralove, 500 05 Hradec Kralove, Czech Republic
* Author to whom correspondence should be addressed.
Abstract: Quinaldine derivatives were obtained, some of them by means of novel synthetic methods. All the compounds were tested for their antifungal activity. The synthetic approach, analytical and spectroscopic data of all newly synthesized compounds are presented. Structure-activity relationships among the chemical structure, the physical properties and the biological activity of the evaluated compounds are discussed. Brome derivatives of 2-methylquinolin-8-ol possessed the highest antifungal activity against all evaluated fungal strains.
Keywords: Quinaldine derivatives; Antifungal evaluation; Structure-activity relationships
Introduction
The compounds based on quinoline ring have been clinically used as antibacterial, antifungal and antiprotozoic drugs [1] and further compounds have been synthesized and evaluated for their antiseptic activities [2,3]. Our previous studies described syntheses and biological evaluations of pyrazine-2-carboxylic acid derivatives or pyridine-4-carboxylic acid derivatives [4-16].
This presented study is a follow-up paper to the previous articles and deals with the synthesis of quinaldine derivatives as substituted bicyclical pyridine derivatives and searched for the structure-activity relationships in the mentioned first series of substituted quinaldine derivatives.
The quinoline is related to pyridine in the same way as naphthalene. The π-electron systems in quinoline and naphthalene are similar in so far as each has 10π-electrons of which two are shared by each ring; giving effectively 6π-electrons in each ring, see Fig. 1. In the heterocyclic compounds the π-electrons can be considered to be donated one from the nitrogen atom and one each carbon atom, see Fig. 2. These electrons are responsible for the weakly basic character of the quinoline (quinoline pKa: 4.94; pyridine pKa: 5.20) [17].
Figure 1: Quinoline can be regarded alternatively as resonance hybrids of various hypothetical canonical forms.
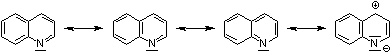 |
Figure 2: Calculation of π-electrons density in the quinoline and pyridine rings has been carried out by the self-consistent field M.O. method [18].
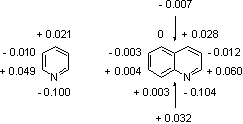 |
Of the seven possible monohydroxyquinolines only those with hydroxyl groups in the 3-, 5-, 6-, 7- and 8- positions are true phenols (8-hydroxyquinoline possess an intramolecular hydrogen bond). The basicities of these hydroxyquinolines are all similar, and slightly higher than that of quinoline itself; this aspect can be ascribed to electron donation by the OH groups [17].
Figure 3: Phenolic monohydroxyquinolines and IR spectra (νmax).
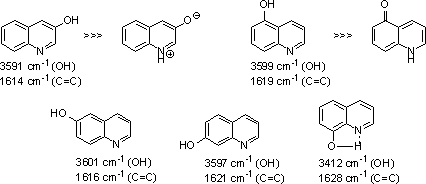 |
2- and 4-Hydroxyquinolines are normally obtained as their tautomers, the 2- and 4-quinoinones. The basicities (pKa: -0.71) of both tautomeric mixtures are different from quinoline and true hydroxyquinolines [17].
Figure 4: 2- and 4-Hydroxyquinolines tautomerism and IR spectra (νmax).
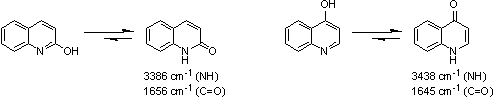 |
Results and Discussion
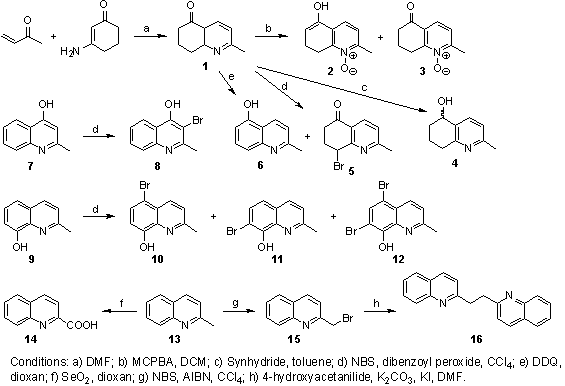
The compounds synthesis is shown in Scheme 1. We described new/more advantageous preparations of some compounds.
The main starting material 1 was obtained by means of condensation of but-3-en-2-one with 3-aminocyclohex-2-enone [19]. Compounds 2 and 3 were generated via N-oxidation with m-chloroperoxybenzoic acid (MCPBA) in dichloromethane (DCM). Ketone 1 was reduced with Synhydride (70% solution of bis(2-methoxyethoxy)dihydroalanate sodium in toluene) that gave 60% yield of racemic secondary alcohol 4. Radical oxidative bromination of ketone 1 using N-bromosuccinimide (NBS) yielded compounds 5 (5%) and 6 (71%), nevertheless compound 6 was obtained also by means of oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in 83% yield. Literature described compound 6 preparation from 2-methylquinolin-5-ylamine using NaNO2, CuNO3, CuO in 32% yield [20] or from 5-amino-2,4-dibromophenol and but-2-enal over multistep synthesis in 61% yield [21].
Compounds 7, 9 and 13 were further starting materials and all were oxidatively brominated using NBS and dibenzoyl peroxide (compound 13 with NBS and 2,2'-azobisisobutyronitrile (AIBN) [22]) to give compounds 8, 10-12, 15. Preparations of compounds 8, 10-12 were described using water hydrobromic acid or by means of condensation of 2-amino-4-bromophenol with but-2-enal or bromination using NBS under homogeneous catalysis by sulphuric acid or copper(II)chelates from different precursors [23-26].
Quinaldine (13) was also oxidized using SeO2 to acid 14 in 57%. An interesting reaction is dimerization of compound 15. Product 16 was generated in 42% yield and quinaldine (13) in 49% yield via reduction with 4-hydroxyacetanilide. The latter was oxidized to N-acetyl-p-benzoquinoneimine, which was isolated as a yellow crystalline compound with Mp: 109-111 °C; [27] Mp: 107-109 °C.
The melting points, yields, as well as the CHN analyses, IR, 1H and 13C NMR spectral data for all the newly prepared compounds 2-5, 16 are given in Experimental. Calculated log P values are shown in Table 1.
Scheme 1: Quinaldine derivatives synthesis.
 |
Sixteen studied compounds were tested for their antifungal activity, but only six compounds 2, 4, 10-12, 16 possess interesting inhibitory activities, see Table 1.
The biological activity showed linear increase with increasing lipophilicity of the compounds within this series. The most active compound 12 possesses similar lipophilicity to the standard ketoconazole. The activity decreases with higher lipophilicity that the latter possesses.
The results from this observation have exposed on the importance of phenolic moiety or hydroxylic group, which can be conjugated with heteroaromatic ring, and bromine for sufficient lipophilicity value for antifungal activity. Compound 8 was not tested due to low solubility in the testing medium (compound 8 occurs as quinone likely, according to Fig. 4), whereas isomers 10-12 possess the necessary solubility and high antifungal activity against all evaluated fungal strains. Regioisomer 11 (bromine in C(7) of quinaldine) showed more activity than compound 10 (bromine in C(5) of quinaldine). An interesting activity showed N-oxide 2. Alcohol 4 possesses higher activities than its aromatic analogues 6, 7, 9; probably due to low solubility of the latter compounds in the testing medium.
Experimental
General
All solvents used for the synthesis were of analytical grade. The solvents were dried and freshly distilled under an argon atmosphere. 2-Methylquinoline, 2-methylquinolin-4-ol and 2-methylquinolin-8-ol were purchased from Sigma-Aldrich. Kieselgel 60, 0.040-0.063 mm (Merck, Germany) was used for flash chromatography (FC) and a silica gel was impregnated TEA. TLC was performed on Silufol UV 254 plates (Kavalier, Votice, Czech Republic) and plates were impregnated TEA. The plates were illuminated under UV (254 nm). Melting points were determined on Boetius PHMK 05 (VEB Kombinat Nagema, Radebeul, Germany) and are uncorrected. Elemental analyses were carried out on an automatic microanalyser EA1110CE (CE Instruments, Milano, Italy). Infrared spectra were recorded with neat oils (for non-crystalline materials) and in KBr pellets (for crystalline materials) on an IR-spectrometer Nicolet Impact 400. 1H and 13C NMR Spectra were recorded on a Varian Mercury - Vx BB 300 (299.95 MHz for 1H and 75.43 MHz for 13C), Bruker Comp. (Karlsruhe, Germany). Chemical shifts are given relative to internal Si(CH3)4.
Log P values were computed using the program CS ChemOffice Ultra ver. 7.0 (CambridgeSoft, Cambridge MA, USA).
Synthesis
2-Methyl-7,8-dihydro-6H-quinolin-5-one (1). 3-Aminocyclohex-2-enone (5.6 g, 50.0 mmol) and but-3-en-2-one (7.0 g, 100.0 mmol) were refluxed in DMF for 1 h. Then the solvent was removed at reduced pressure. FC (Et2O/petroleum ether 6:1) gave a colourless oil. Yield: 4.2 g (52%). RF: 0.38 (Et2O/petroleum ether 6:1). 1H and 13C NMR spectra identical with reference [19].
5-Hydroxy-2-methyl-7,8-dihydroquinolin-1-oxide (2). Compound 1 (2.0 g, 12.6 mmol) was dissolved in dry DCM and 50% MCPBA (6.0 g) was added. The mixture was stirred under room temperature for 16 h. The reaction mixture was transferred to a separation funnel and extracted with EtO2 and saturated solution of Na2CO3. The combined organic extracts were dried over anhydrous MgSO4 and filtered. The solvent was removed at reduced pressure. FC (acetone/petroleum ether 3:2) gave a yellow crystalline compound. Yield: 1.2 g (55%). Mp: 152-154 °C. RF: 0.33 (acetone/petroleum ether 3:2). For C10H11NO2 (177.20) calculated: 67.78% C, 6.26% H, 7.90% N; found: 67.75% C, 6.26% H, 7.87% N. IR (KBr), cm-1: 3436, 1186 (OH), 3027 (=CH-), 2958 (CH3), 3085, 1567, 1103 (pyridine), 1442 (CH2), 1259 (N-O). 1H NMR (300 MHz, CDCl3), δ: 7.83 (d, 1H, J=8.25 Hz, H4), 7.36 (d, 1H, J=8.24 Hz, H3), 4.61 (dd, 1H, J=6.32 Hz, J=4.39 Hz, CH), 3.42-3.35 (m, 2H, CH2), 2.61 (s, 3H, CH3), and 2.60-2.52 (m, 2H, CH2). 13C NMR (75 MHz, CDCl3), δ: 188.7, 154.3, 152.7, 127.2, 124.3, 123.7, 57.4, 29.0, 21.6, and 18.9.
2-Methyl-5-oxy-5,6,7,8-tetrahydroquinolin-1-oxide (3). See compound 2 for conditions. A yellow crystalline compound. Yield: 0.5 g (23%). Mp: 74.5-76 °C. RF: 0.23 (acetone/petroleum ether 3:2). For C10H11NO2 (177.20) calculated: 67.78% C, 6.26% H, 7.90% N; found: 67.81% C, 6.23% H, 7.86% N. IR (KBr), cm-1: 2957 (CH3), 3086, 1566, 1109 (pyridine), 1696 (C=O), 1441 (CH2), 1257 (N-O). 1H NMR (300 MHz, CDCl3), δ: 7.79 (d, 1H, J=7.83 Hz, H4), 7.29 (d, 1H, J=7.83 Hz, H3), 3.25 (t, 2H, J=6.46 Hz, CH2), 2.66 (t, 2H, J=6.46 Hz, CH2), 2.59 (s, 3H, CH3), and 2.27-2.15 (m, 2H, CH2). 13C NMR (75 MHz, CDCl3), δ: 195.9, 154.3, 153.5, 129.3, 123.7, 122.9, 37.4, 24.5, 20.3, and 18.9.
2-Methyl-5,6,7,8-tetrahydroquinolin-5-ol (4). Ketone 1 (1.2 g, 0.8 mmol) was dissolved in dry toluene (30 ml) and Synhydride (70% solution in toluene, 10 ml) was added drop wise with stirring. The mixture was refluxed for 2 h. After cooling, 15% aqueous HCl (30 ml) was added. The mixture was extracted with Et2O, the combined Et2O extracts were dried over anhydrous MgSO4, filtered and the solvent was removed under reduced pressure. FC (Et2O/MeOH 4:1) gave a light yellow crystalline compound. Yield: 0.5 g (60%). RF: 0.17 (Et2O). Mp: 95-97 °C. For C10H13NO (163.22) calculated: 73.59% C, 8.03% H, 8.58% N; found: 73.64% C, 8.06% H, 8.57% N. IR (KBr), cm-1: 3416, 1091 (OH), 2934 (CH3), 3056, 1592, 1101 (pyridine), 1465 (CH2). 1H NMR (300 MHz, DMSO-d6), δ: 8.60 (d, 1H, J=7.93 Hz, H4), 7.03 (d, 1H, J=7.93 Hz, H3), 4.37 (dd, 1H, J=5.00 Hz, J=5.00 Hz, CH), 3.46-3.39 (m, 2H, CH2), 2.73-2.67 (m, 2H, CH2), 2.50 (s, 3H, CH3), and 2.19-2.08 (m, 2H, CH2). 13C NMR (75 MHz, DMSO-d6), δ: 156.2, 155.9, 136.6, 132.6, 120.7, 66.1, 41.1, 32.1, 24.1, and 21.7.
8-Bromo-2-methyl-7,8-dihydro-6H-quinolin-5-one (5). A solution of 1 (0.5 g, 3.1 mmol), NBS (0.6 g, 3.1 mmol), and dibenzoyl peroxide (0.016 g) in dry CCl4 was refluxed for 8 h under argon. The mixture was cooled in an ice bath, filtered, and the filtrate was concentrated in vacuum. FC (Et2O/petroleum ether 2:3) gave a yellow crystalline compound. Yield: 0.03 g (5%). RF: 0.70 (Et2O). Mp: 107-109 °C. For C10H10BrNO (240.10) calculated: 50.02% C, 4.20% H, 5.83% N; found: 50.07% C, 4.26% H, 5.81% N. IR (KBr), cm-1: 2938 (CH3), 3064, 1584, 1101 (pyridine), 1703 (C=O), 1463 (CH2), 611 (C-Br). 1H NMR (300 MHz, CDCl3), δ: 8.18 (d, 1H, J=7.85 Hz, H4), 7.24 (d, 1H, J=7.86 Hz, H3), 5.60 (t, 1H, J=7.25 Hz, CH), 3.22-3.09 (m, 2H, CH2), 2.85-2.70 (m, 2H, CH2), and 2.60 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3), δ: 195.9, 164.3, 160.4, 135.7, 124.3, 121.7, 49.3, 34.1, 30.9, and 25.0.
2-Methylquinolin-5-ol (6). Method A: See compound 5 for conditions. Yield: 0.4 g (71%). Method B: Ketone 1 (1.0 g, 6.3 mmol) and DDQ (1.4 g, 6.3 mmol) were dissolved in dry dioxan and stirred for 24 h under argon. Then the mixture was concentrated in vacuum and saturated solution of Na2CO3 was added. The mixture was extracted with Et2O, the combined Et2O extracts were dried over anhydrous MgSO4, filtered and the solvent was removed under reduced pressure. FC (Et2O/petroleum ether 5:1). Yield: 0.8 g (83%). A white crystalline compound. RF: 0.25 (Et2O). Mp: 227-229 °C; [20] Mp: 230-232 °C.
3-Bromo-2-methylquinolin-4-ol (8). Compound 7 bromination. See compound 5 for conditions. The crude product was purified by crystallization from EtOH/H2O, and a white crystalline compound was obtained. Yield: 0.8 g (98%). RF: 0.55 (acetone/toluene 4:1). Mp: 322-324 °C; [24] Mp: 326 °C.
5-Bromo-2-methylquinolin-8-ol (10). Compound 9 bromination. See compound 5 for conditions. FC (Et2O/petroleum ether 1:6) gave a white crystalline compound. Yield: 0.03 g (4%). RF: 0.32 (Et2O/petroleum ether 1:6). Mp: 68.5-70.0 °C; [23] Mp: 68 °C.
7-Bromo-2-methylquinolin-8-ol (11). Compound 9 bromination. See compound 5 for conditions. A white crystalline compound. Yield: 0.2 g (20%). RF: 0.23 (Et2O/petroleum ether 1:6). Mp: 137-139 °C; [25] Mp: 134-136 °C.
5,7-Dibromo-2-methylquinolin-8-ol (12). Compound 9 bromination. See compound 5 for conditions. A white crystalline compound. Yield: 0.1 g (11%). RF: 0.12 (Et2O/petroleum ether 1:6). Mp: 124-126 °C; [23] Mp: 125 °C.
2-Bromomethylquinoline (14). 2-Methylquinoline (1.0 g, 6.9 mmol) was dissolved in dry CCl4 and refluxed in the presence of NBS (1.3 g, 6.9 mmol) and AIBN (0.014 g). After cooling, the solution was filtered, evaporated and the residue was washed with MeOH to give a yellow crystalline compound. Yield: 0.9 g (55%). RF: 0.57 (acetone/petroleum ether 1:4). Mp: 67-68 °C; [28] Mp: 63-64 °C.
Quinoline-2-carboxylic acid (15). A mixture of 13 (1.0 g, 7.0 mmol) and SeO2 (2.5 g, 21.0 mmol) in dioxan was refluxed for 8 h. After cooling, the solution was evaporated and the residue was extracted with EtOAc and H2O. The combined organic extracts were dried over anhydrous MgSO4, filtered and the solvent was removed under reduced pressure. The crude product was purified by crystallization from EtOH/H2O and then from H2O only, and a light yellow crystalline compound was obtained. Yield: 0.7 g (57%). RF: 0.47 (MeOH/isopropylalcohol 1:1). Mp: 158-159 °C; [29] Mp: 155 °C.
2-(2-quinolin-2-ylethyl)quinoline (16). A stirred mixture of 4-hydroxyacetanilide (0.5 g, 3.5 mmol), 2-bromomethylquinoline (0.8 g, 3.5 mmol), anhydrous K2CO3 (3.0 g), KI (0.1 g) and DMF (50 ml) was refluxed for 3 h. The hot mixture was filtered, the filtration cake was washed with boiling DMF, and the solution was evaporated under reduced pressure. FC (acetone/petroleum ether 1:4) was obtained a yellow crystalline compound. Yield: 0.4 g (42%). RF: 0.35 (acetone/petroleum ether 1:4). Mp: 159-161 °C; [30] Mp: 163 °C.
In vitro antifungal susceptibility testing
The broth microdilution test [31, 32] was used for the assessment of in vitro antifungal activity of the synthesized compounds against Candida albicans ATCC 44859 (CA), Candida tropicalis 156 (CT), Candida krusei E28 (CK), Candida glabrata 20/I (CG), Trichosporon beigelii 1188 (TB), Aspergillus fumigatus 231 (AF), Absidia corymbifera 272 (AC), and Trichophyton mentagrophytes 445 (TM) in comparison with ketoconazole. The procedure was performed with twofold compounds in RPMI 1640 buffered to pH 7.0 with 0.165 mol of 3-morpholino-propane-1-sulfonic acid. The final concentrations of the compounds ranged from 1000 to 0.975 µmol . dm-3. Drug-free controls were included. The MICs were determined after 24 and 48 h of static incubation at 35 °C. With Trichophyton mentagrophytes, the final MICs were determined after 72 and 120 h of incubation.
Table 1: Antifungal activity and lipophilicity (calculated log P) of compounds 2, 4, 10-12, 16 in comparison with standard ketoconazole (KET).
| Compound | Log P | MIC/EC80 (µmol . dm-3) | |||||||
| CA | CT | CK | CG | TB | AF | AC | TM | ||
| 24h 48h |
24h 48h |
24h 48h |
24h 48h |
24h 48h |
24h 48h |
24h 48h |
72h 120h |
||
| 2 | a | b | b | b | b | b | b | b | 31.25 31.25 |
| 4 | 1.48 | 125 >125 |
125 >125 |
125 >125 |
125 >125 |
125 >125 |
125 >125 |
125 >125 |
125 >125 |
| 10 | 3.52 | 3.91 7.81 |
31.25 31.25 |
7.81 7.81 |
31.25 31.25 |
15.63 62.5 |
15.63 31.25 |
31.25 62.5 |
31.25 62.5 |
| 11 | 3.52 | 3.91 3.91 |
15.63 31.25 |
3.91 7.81 |
15.63 15.63 |
31.25 31.25 |
15.63 15.63 |
15.63 15.63 |
15.63 15.63 |
| 12 | 4.09 | <0.45 0.9 |
7.81 15.63 |
7.81 15.63 |
7.81 15.63 |
31.25 62.5 |
15.63 15.63 |
62.5 62.5 |
7.81 15.63 |
| 16 | 5.28 | 125 >125 |
125 >125 |
125 >125 |
125 >125 |
125 >125 |
125 >125 |
125 >125 |
125 >125 |
| KET | 4.01 | <0.24 <0.24 |
1.95 3.91 |
0.98 1.95 |
0.49 1.95 |
<0.24 <0.24 |
7.81 7.81 |
31.25 31.25 |
0.98 1.95 |
a not computed due to charges; b no interesting activity.
Acknowledgements. The Ministry of Education of the Czech Republic (Projects No. MSM 111600001 and No. MSM 111600002) supported this study. We also thank Mrs. V. Hronova, Mrs. I. Vencovska and Mr. T. Vojtisek from the Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Czech Republic.
References