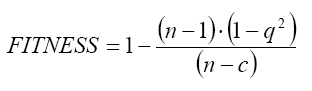2- MARCH-INSIDE Methodology.
2.1- General Definitions:
A precise definition of the descriptors generated by this methodology can be found in several reports of its application in the study of several biological properties 13-19. Briefly we can say that MARCH-INSIDE methodology considers as states of the MCH the electrons layers of any atom in the molecule. The method uses as source of molecular descriptor the П1 matrix (the one-step electron-transition stochastic matrix) built up as a squared matrix nxn (n number of atoms in the molecule) whose elements (pij) are calculated as the ratio between the withdrawing of the jth atom and the sum over all the atoms covalently linked to the ith atom. Also a new matrix, AПk matrix, can be defined as the product of a 1xn vector (AП0) whose elements (Aπk(j)) are calculated in the similar way as pij but the sum is carried out now over the all the atoms in the molecule and the kП which is the k-th power of 1П matrix. These matrices van then be used to generate three families of molecular descriptors: Self return Probabilities (SRπk): Can be defined as the trace (sum over the pii values) of the k-th power of the 1П matrix.

Codify the attraction of an atom or group of atoms for its electrons (the electrons that were at the atom or the given group of atoms in the time t0) at any time tk located at k-th steps away or less. Absolute Probabilities (Absπk(j)): Codify the attraction of the j-th atom over any electron in the molecule at any time tk after traveling by different paths of less than k steps.
Electronic Delocalization Entropy (Θk):

It describes the entropy involved in the electron attraction at lest k steps beginning with the j atom.
2.2- Descriptors Calculation and Physico-Chemical properties of amino acids:
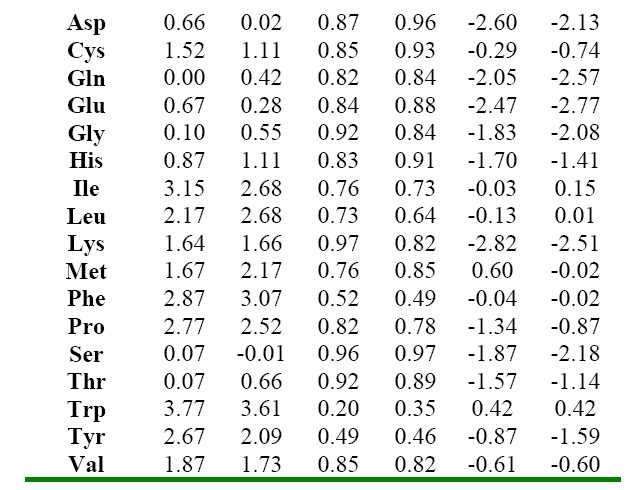
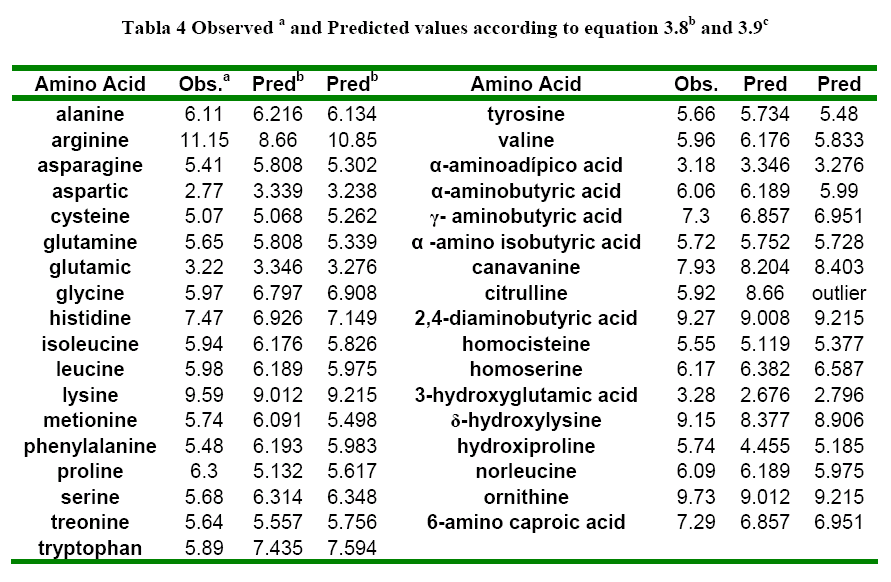
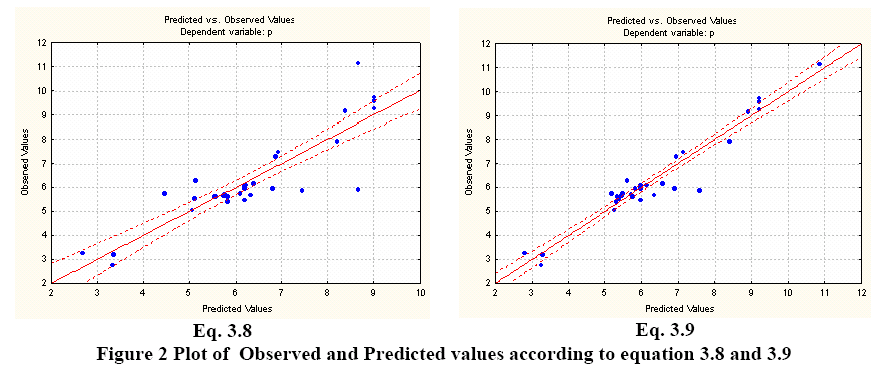
The molecular descriptors (SRπk Θ k Absπk) were calculated with the experimental software MARCH-INSIDE version 2.0 20. The chemical structure is directly introduced by using the Draw Mode of the software. The structure can then be saved and is possible to select the Calculus Mode of the software and to obtain the first 10th –order local (as desired) and total molecular descriptors. In these case the total descriptors as well as the local descriptors over the residue (R), Heteroatom, and Hydrogen directly bonded to Heteroatom were calculated. The Physico-Chemical properties of amino acids were extracted from the reports of Hellberg 6 in the first z-scale report and Liu 2 with a larger data including synthetic amino acids (up to 35) for the prediction of the pI. The Table 1 shows the properties modeled in this study.

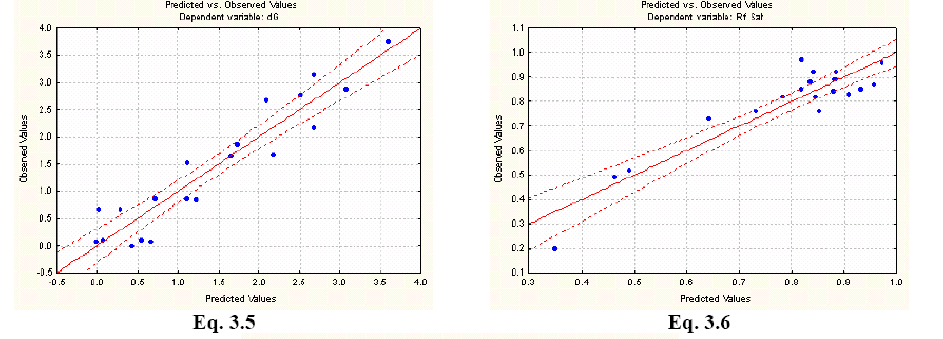
The STATISTICA software 21 was used to develop the Multiple Linear Regression. Several statistical parameters were taken into account to asses the statistical quality of the models: Correlation coefficient (R2), Standard Error of Determination (s), Fischer ratio (F), Cross Validation Regression Coeficient (R2cv or q2).