8th International Electronic Conference on Synthetic Organic Chemistry. ECSOC-8. 1-30 November 2004. http://www.lugo.usc.es/~qoseijas/ECSOC-8/
[A026]
Synthesis of asymmetrically meso-substituted porphyrins bearing amino groups as potential cationic photodynamic agents
ABSTRACT. Novel asymmetrically meso-substituted porphyrins bearing amino groups have been synthesized as precursors of cationic photodynamic agents. The amphiphilic character of porphyrins was increased by the presence of a high lipophilic trifluoromethyl group. Different patterns were obtained from meso-4-[(3-N,N-dimethylaminopropoxy)phenyl]dipyrromethane 1, which was formed by the condensation of 4-(3-N,N-dimethylaminopropoxy)benzaldehyde with a large excess of pyrrole. This reaction takes place at high temperature with a yield of 59 %. To obtain porphyrins, dipyrromethane 1 was condensed with aldehydes in the presence of trifluoroacetic acid (TFA) under different conditions. First, 1 was reacts with 4-(3-N,N-dimethylaminopropoxy)benzaldehyde in dichloromethane catalyzed by TFA (~4 times TFA/1 molar ratio) to obtain 6.2 % of 5,10,15,20-tetrakis(4-[3-N,N-dimethylaminopropoxy]phenyl)porphyrin (A4-porphyrin). Under similar conditions, reaction of 1 with 4-(trifluoromethyl)benzaldehyde produces 5,15-di(4-[3-N,N-dimethylaminopropoxy]phenyl)-10,20-di(4-trifluoromethylphenyl)porphyrin (A2B2-porphyrin) with 4.8 % yield. Also, this procedure yields a mixture of porphyrins, which were formed due to acidolysis of 1. When minor amount of TFA was used in acetonitrile the yield of A2B2-porphyrin was very poor (~0.4 %). On the other hand, condensation of 1 with 4-trifluoromethylbenzaldehyde and 4-(3-N,N-dimethylaminopropoxy)benzaldehyde catalyzed by TFA (~2 times TFA/1 molar ratio) in acetonitrile yields 9.3 % of 5-(4-trifluoromethylphenyl)-10,15,20-tris(4-[3-N,N-dimethylaminopropoxy]phenyl) porphyrin (A3B-porphyrin). Also, A2B2 and A4 porphyrins were isolated with 6.0 and 2.0 %, respectively. Finally, exhaustive methylation of amino porphyrins produce cationic sensitizers (>94 % yields). In these porphyrins, the cationic centers are isolated from the porphyrin ring by a propoxy bridge, which also provides a higher mobility of the charge facilitating the interaction with the outer membrane of the Gram-negative bacteria. These amphiphilic cationic porphyrins are promising photosensitizers with potential applications in bacteria inactivation by photodynamic therapy.
Introduction
In recent years, the changing pattern of infectious disease and the emergence of antibiotic resistant bacterial strains have made it necessary to find new approaches for the treatment of bacterial infections [[1],[2]]. Photodynamic inactivation (PDI) is one novel alternative for such treatments. The studies show that Gram-positive bacteria are susceptible to the photosensitizing action of a variety of sensitizers. However, negatively charged or neutral sensitizers exhibit a low activity in the photoinactivation of Gram-negative bacteria, whereas, cationic porphyrin derivatives have shown to photoinduce direct inactivation of Gram-negative bacteria. The positive charge on the photosensitizer molecule appears to promote a tight electrostatic interaction with negatively charged sites at the outer surface of the bacterial cells.
The combination of hydrophobic and hydrophilic substituents in the sensitizer structure results in an intramolecular polarity axis, which can facilitate membrane penetration and produces a better accumulation in subcellular compartments, enhancing the effective photosensitization [[3]]. The design of these sensitizers architecture required the formation of asymmetrically meso-substituted porphyrins. These porphyrins containing two different types of meso-substituents can be prepared by a binary mixed aldehyde and pyrrole condensation. However, this approach is statistical in nature and usually six porphyrins are formed [[4]]. The isolate requires slowly chromatographic separation, the yield is very poor and no pure porphyrin is always possible. A convenient approach for the synthesis of meso-substituted trans-porphyrins (A2B2-porphyrins) has been developed from the condensation of dipyrromethane with an aldehyde in a MacDonald-type 2+2 condensation catalyzed by acid [[5],[6]]. Also, condensation of dipyrromethane with a binary mixture of aldehydes was used to obtain meso-substituted porphyrins bearing three identical molecular structures B and one different A (AB3-porphyrins) [[7],[8]]. In these cases, the structure A bears a functional group, which can be used to link the porphyrin with other molecules, while B was used to change the macrocycle properties.
The main problem with the synthesis of porphyrins containing amino groups results from the interaction of an acid catalyst with substrates [[9],[10]]. In such cases, the amount of acid catalyst necessary for the reaction to proceed effectively is difficult to predict.
In this work, combinations of positive charges incorporated at the peripheral position were use to increase the amphiphilic character of the porphyrin structures. This effect could help porphyrin derivatives to pass through or accumulate in biomembranes. In these porphyrins, the cationic centers are isolated from the porphyrin ring by a propoxy bridge and thus the charges have minimal influence on the electron density of the tetrapyrrolic macrocycle. Also, this chain provides a higher mobility of the charge, which could facilitate the interaction with the outer membrane of the Gram-negative bacteria.
Dipyrromethane formation
meso-[4-(3-N,N-Dimethylaminopropoxy)phenyl]dipyrromethane 1 was obtained from a aldehyde and pyrrole mixture at elevated temperature in the absence of acid with appreciable yield of 59 % (Scheme 1). The dipyrromethane 1 was purified by removal the excess of pyrrole under high-vacuum distillation and by flash chromatography on silica gel in a mildly basic medium, using ethyl acetate/triethylamine (TEA) 1%/methanol 5-10% gradient as eluent.
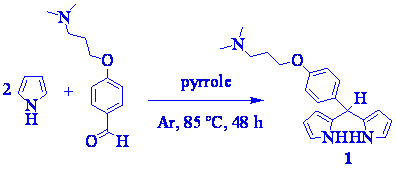
Scheme 1
Synthesis of porphyrin
At first attempt to evaluate the reactivity of dipyrromethane 1, it was reacts under conditions where only one porphyrin is expected to form. Thus, a mixture of 1 (~10 mM) and 4-(3-N,N-dimethylaminopropoxy)benzaldehyde was treated with TFA (~4 times TFA/reactant molar ratio) in dichloromethane for 30 min at room temperature (Scheme 2). After oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), the predictable 5,10,15,20-tetrakis[4-(3-N,N-dimethylaminopropoxy) phenyl]porphyrin (A4) was purified by flash chromatography affording 6.2 % yield.
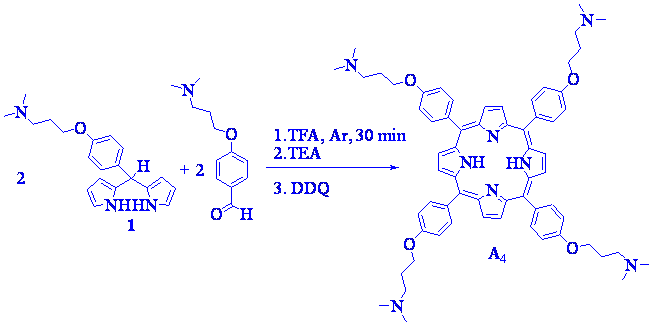
Scheme 2
To obtain porphyrins with different patters of substitution, dipyrromethane 1 was condensed with benzaldehydes under different conditions. First, dipyrromethane 1 (~10 mM) was reacted with 4-(trifluoromethyl)benzaldehyde in 1:1 mol ratio using dichloromethane as solvent (Scheme 3). The reaction was treated with trifluoroacetic acid (TFA) (~4 times TFA/reactant molar ratio) at room temperature for 30 min. This amount of acid was used to neutralize the amine groups and to act as catalyst. After that, the reaction mixture was subject to oxidation with DDQ. The condensation under these conditions produces a mixture of porphyrins, which were separated by flash chromatography with high purity using dichloromethane/methanol/TEA gradient. The first purple band, corresponds to 5,10,15,20-tetrakis(4-trifluoromethylphenyl)porphyrin (B4) yielding 2.8 %. Then, 5-(4-[3-N,N-dimethylaminopropoxy]phenyl)-10,15,20-tris(4-trifluoromethylphenyl)porphyrin (AB3) 5.4 %, 5,15-di(4-[3-N,N-dimethylaminopropoxy]phenyl)-10,20-di(4-trifluoromethylphenyl)porphyrin (A2B2) 4.8 %, 5,10,15-tris[4-(3-N,N-dimethylaminopropoxy)phenyl]-20-(4-trifluoromethylphenyl)porphyrin (A3B) 3.0 % and 5,10,15,20-tetrakis[4-(3-N,N-dimethylaminopropoxy)phenyl] porphyrin (A4) 2.0 %, were sequentially obtained. Therefore, although only one porphyrin is also expected in this experiment, a mixture of porphyrin derivatives with different patters of substitution was obtained. In this case, an excess acid was used to overcome the basicity of the amine group in the dipyrromethane 1. However, this amount of TFA produces acidolysis of dipyrromethane 1, which induces the presence of scrambling in the reaction [9,[11]].
To evaluate the effect produced by the acid, the reaction between dipyrromethane 1 (2.5 mM) and 4-(trifluoromethyl)benzaldehyde (1:1 mol ratio) was also investigate using 3 mM of TFA (1.2 times TFA/1 molar ratio). The condensation was performed at room temperature in acetonitrile as solvent. This more polar solvent was used in the reaction of a dipyrromethane bearing a nitrogen heterocyclic and dipyrrolmethane-dicarbinol [9]. After different reaction times, an aliquot (2 mL) of reaction mixture was treated with DDQ for 2 h and then with 1 mL of methanol. The porphyrin formation was analyzed by fluorescence spectroscopy. The results show that under these conditions no significant amount of porphyrin was observed after 1 h of reaction. The overnight reaction yielded ~0.4 % of porphyrin and a prolongation of the reaction time for 48 h of stirring under Ar atmosphere did not improve the yield.
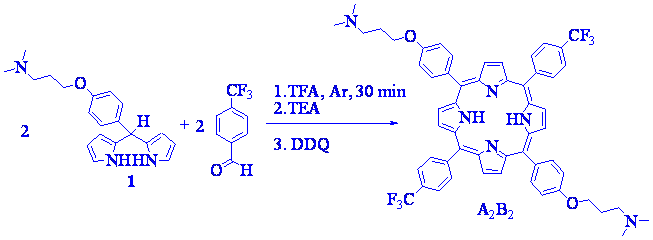
Scheme 3
On the other hand, one way to prepare porphyrin bearing two different types of meso-substituents, such as A3B-porphyrin, could be by a condensation of a binary mixture of aldehyde and dipyrromethane [7]. Thus, and taking into account the previous results, condensation of dipyrromethane 1 (2.5 mM) with 4-trifluoromethylbenzaldehyde and 4-(3-N,N-dimethylaminopropoxy)benzaldehyde (2.5:1:1 molar ratio) was performed using 5 mM of TFA (Scheme 4). The reaction was performed in acetonitrile at room temperature for 2 h. After that, the acid was neutralized with TEA, the volatiles evaporated, dissolution in dichloromethane and treated with DDQ for 3 h. This procedure produce a mixture of the three expected porphyrin i.e. A4, A3B and A2B2 porphyrins, which were isolated by flash chromatography using dichloromethane/methanol/TEA gradient as eluent. Under these conditions, the reaction yields 2.0 % of A4-porphyrin, 9.3 % of A3B-porphyrin and 6.0 % of A2B2-porphyrin. Also, this procedure produces 0.3 % of AB3-porphyrin.
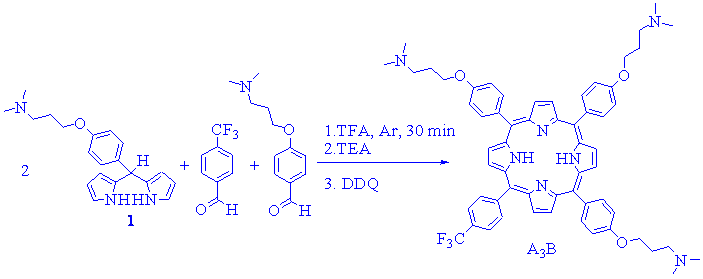 Scheme 4
Scheme 4
Finally, cationic sensitizers were obtained treating the amino porphyrins with methyl iodide for 72 h at reflux in acetone (Scheme 5). The exhaustive methylation produces 5,10,15,20-tetrakis[4-(3-N,N,N-trimethylammoniumpropoxy)phenyl]porphyrin iodide (A44+), 5,10,15-tris[4-(3-N,N,N-trimethylammoniumpropoxy) phenyl]-20-(4-trifluoromethylphenyl)porphyrin iodide (A3B3+), 5,15-di[4-(3-N,N,N-trimethylammoniumpropoxy) phenyl]-10,20-di(4-trifluoromethylphenyl) porphyrin iodide (A2B22+) and 5-[4-(3-N,N,N-trimethylammoniumpropoxy) phenyl]-10,15,20-tris(4-trifluoromethylphenyl)porphyrin (AB3+) with > 94 % yields.
All the products were characterized by absorption, fluorescence, MS and 1HNMR spectra.

Scheme 5
Conclusions
This work shows that direct condensation of aldehyde with excess of pyrrole at high temperature is possible to form dipyrromethane 1 with appreciable yield (59 %). This result enlarges the scope of this reaction to aromatic aldehyde substituted by an aliphatic chain bearing an amine group. The second step to obtain porphyrin involves the condensation of dipyrromethane 1 with different mixture of aldehydes catalyzed by TFA. Also, in these cases the presence of amine groups make difficult to estimate the amount of acid catalyst necessary for the reaction to proceed effectively. Therefore, we tried different condition of reaction. First, dipyrromethane 1 was reacts with 4-(3-N,N-dimethylaminopropoxy)benzaldehyde in dichloromethane catalyzed by TFA (~4 times TFA/1 molar ratio) to obtain 6.2 % of A4-porphyrin. Under similar conditions, reaction of dipyrromethane 1 with 4-(trifluoromethyl)benzaldehyde produces a mixture of porphyrins, which were formed due to acidolysis of dipyrromethane 1. On the other hand, when minor amount of TFA (~1.2 times TFA/1 molar ratio) was used in acetonitrile, the yield of A2B2-porphyrin was very poor (~0.4 %) still after 48 h of reaction. Thus, when a large excess of acid is used the reaction is accompanied by an increase in the scrambling whereas a lower amount of TFA produces a diminishing in the porphyrin yield still after long reaction time. Taking into account these results, to synthesize A3B-porphyrin, dipyrromethane 1 was condensed with a binary mixture of 4-trifluoromethylbenzaldehyde and 4-(3-N,N-dimethylaminopropoxy)benzaldehyde catalyzed by TFA (~2 times TFA/1 molar ratio) in acetonitrile. This condition appears to be the more appropriated to obtain A3B-porphyrin with 9.3 % yield. Although this yield is lower than those obtained for the formation of other porphyrin with the amino groups protected (~15-17 %) [7,8], the present pathway diminishes the number of reaction steps.
Finally, to obtain the porphyrin targets of these work, exhaustive methylation of amino porphyrins were performed to obtain cationic sensitizers (>94 % yields). The amphiphilic character of porphyrins was increased by the presence of a high lipophilic trifluoromethyl group. In these porphyrins, the cationic centers are isolated from the porphyrin ring by a propoxy bridge, which also provides a higher mobility of the charge facilitating the interaction with the outer membrane of the Gram-negative bacteria. These amphiphilic cationic porphyrins offer a promising molecular architecture for photosensitizer agents with potential applications in bacteria inactivation by photodynamic treatment. Studies of photosensitization in vitro, using Gram-negative bacteria Escherichia coli, are presently in progress in our laboratory.
Acknowledgements. Authors are grateful to Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) of Argentina and Fundación Antorchas for financial support. E.N.D. is Scientific Members of CONICET. D.A.C. thanks Fundación Antorchas for a research fellowship.
References