|
Molbank 2006, M464 |
A one-step synthesis of pyrazolone
Department of
Drug Synthesis,
Althanstrasse 14, 1090
Phone:
+43-1-4277-55634, e-mail: [email protected]
*Author to whom
correspondence should be addressed
Received:
Keywords: Pyrazolone, cyclization, NMR
spectroscopy, tautomerism.
Abstract: The fully unsubstituted pyrazolone (=
2-pyrazolin-5-one, which is tautomer to 1H-pyrazol-3-ol and 1H-pyrazol-5-ol) was prepared from hydrazine hydrate and methyl (2E)-3-methoxyacrylate in almost
quantitative yield. Detailed spectroscopic data (1H NMR, 13C
NMR, 15N NMR, MS) for this compound are presented.
Substituted
2-pyrazolin-5-ones play an important role as substructures of numerous
pharmaceuticals, agrochemicals, dyes, pigments, as well as chelating agents and
thus attract remarkable attention [1,2].
Recently,
we investigated the synthesis of some N1-unsubstituted pyrazolones
by use of the PMB (p-methoxybenzyl) protecting group [3,4].
Although this substituent proved to be conveniently
removable from various 4-substituted pyrazolones upon
treatment with refluxing trifluoroacetic acid only
poor results were obtained when the parent 1-PMB-pyrazolone (=
2-(4-methoxybenzyl)-2,4-dihydro-3H-pyrazol-3-one [3]) was subjected to these conditions. Even
prolonged heating (1 week instead of 1 day) did not effect full deprotection (1 day ~ 15%, 2 days ~ 35%, 7 days ~ 75%;
monitored by mean of 1H NMR). Hence, there is need for other and
more suitable methods for the synthesis of the unsubstituted
pyrazolone 1.
With
respect to the fact that other hitherto described syntheses of 1 are characterized by multi-step
procedures and/or low yields [5,6], we report here an almost quantitative
one-step preparation of the fully unsubstituted pyrazolone system from hydrazine hydrate and methyl (2E)-3-methoxyacrylate following an
already known procedure for the synthesis of 1-alkyl pyrazolones
[7] (Scheme 1).
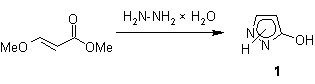
Scheme 1. One-step procedure for the
preparation of Ī«pyrazoloneĪ» 1.
A
considerable number of studies deal with the prototropic
tautomerism of pyrazolones
[8].
Determination of the tautomeric composition of compound 1 is quite challenging as eight possible tautomeric forms have to be considered. This may also be a reason why in the Chemical Abstracts Service (CAS) references are cited for all possible tautomeric forms of compound 1 (Figure 1) except for form E. From the signal multiplicities in the carbon NMR spectra tautomeric forms B, C, F, G, and H can be excluded. Moreover, the 15N NMR chemical shifts found for compound 1 (©C126.5 ppm and ©C192.0 ppm) rule out form A, as for this tautomer a much smaller chemical shift for the =CH©CNH©C atom has to be expected (for instance, in phenazone ©C 1,5-dimethyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-one ©C which is structurally related to form A this atom exhibits a chemical shift of ©C245.1 ppm [9]). The differentiation between forms D and E is not a trivial task. However, from comparison of the 13C chemical shifts, the 15N chemical shifts, the 3J(H,H) coupling constants, and the different 13C,1H-coupling constants of 1 with those of the corresponding N-phenyl analogues (1-phenyl-1H-pyrazol-3-ol, 1-phenyl-1H-pyrazol-5-ol) [10,11] we assume that form D is predominating in DMSO-d6 solution. Nevertheless an additional contribution of other isomers (in minor amounts) can not be ruled out.

Figure 1. Possible tautomeric
forms of Ī«pyrazoloneĪ» 1.
Compound 1: Under stirring,
to a solution of 5.81 g (50 mmol) of methyl (2E)-3-methoxyacrylate in methanol (5 mL) was hydrazine hydrate (2.75 g, 55 mmol)
added and the mixture was refluxed for 1h. Evaporation under reduced pressure
to dryness gave 4.13 g (98%) of a slightly yellowish powder, pure according to 1H
NMR spectroscopy.
Melting point: 160©C162 ĪŃC,
crystal modifications starting at ~140 ĪŃC, (lit. [12] 162©C164 ĪŃC).
1H-NMR (300 MHz,
DMSO-d6, 28 ĪŃC, numbering
for 1H-pyrazol-3-ol = form D) [13]: ”─= 9.82 (br
s, 2H, XH); 7.33 (d, 3J(H5,H4)=
2.3 Hz, 1H, H5); 5.43 (d, 3J(H4,H5)=
2.3 Hz, 1H, H4).
13C-NMR (75 MHz,
DMSO-d6, 28 ĪŃC, numbering
for 1H-pyrazol-3-ol = form D) [13]: ”─= 161.0 (C3, 2J(C3,H4)= 3.4 Hz, 3J(C3,H5)= 9.2 Hz); 130.1 (C5,
1J = 184.0 Hz, 2J(C5,H4)= 8.2 Hz); 89.3 (C4, 1J = 175.6 Hz, 2J(C4,H5)= 8.7 Hz).
15N-NMR (50 MHz,
DMSO-d6, 294 K) [14]: ”─=
©C126.5; ©C192.0.
MS (m/z, %)
[15]: 84 (M+, 100); 55 (24).
Elemental Analysis:
Calculated for C3H4N2O
(84.08): C, 42.86%; H, 4.80%; N, 33.32%. Found: C, 42.75%; H, 4.65%; N, 33.15%.
References
and Notes:
1. J. Elguero, In 'Comprehensive Heterocyclic Chemistry: Pyrazoles and their Benzo
Derivatives', Vol. 5; A. R. Katritzky and C. W. Rees,
Eds., Pergamon Press, Oxford, 1984, 167©C303.
2. Stanovnik, B.; Svete, J. Product
class 1: Pyrazoles. Science of Synthesis 2002, 12, 15©C225.
3. Eller, G.
A.; Holzer, W. Heterocycles 2004, 63, 2537©C2555.
4. Becker, W.; Eller, G. A.; Holzer, W. Synthesis 2005, 2583©C2589.
5. Testa, E.; Fontanella, L. Farmaco 1971, 26,
1017©C35.
6. Dorn, H.; Zubek, A. J. Prakt. Chem. 1971,
313, 1118©C24.
7. Maywald, V.; Steinmetz, A.; Rack, M.; Gotz, N.;
Gotz, R.; Henkelmann, J.; Becker, H.; Aiscar Bayeto, PCT Int. Appl. WO 0031042 A2 2000 (Chem. Abstr., 2000, 133, 4655).
8. Holzer, W.; Hallak, L. Heterocycles 2004, 63, 1311©C1334, and references cited
therein.
9. Cizmarik, J.; Lycka, A. Pharmazie 1988,
43, 794©C795.
10. Holzer, W.; Kautsch, C.; Laggner, C.; Claramunt, R. M.; Perez-Torralba,
M.; Alkorta, I.; Elguero,
J. Tetrahedron 2004, 60, 6791©C6805.
11. Sackus, A.; Holzer, W. manuscript in preparation.
12. Lingens, F.; Schneider-Bernloehr, H. Liebigs Ann. Chem. 1965, 686, 134©C144.
13. The
spectrum was obtained on a Varian UnityPlus 300
spectrometer (299.95 MHz for 1H, 75.43 MHz for 13C). The
center of the solvent signal was used as an internal standard which was related
to TMS with ”─ 2.49 ppm (1H NMR) and ”─ 39.5
ppm (13C NMR).
14. The
spectrum was obtained on a Bruker Avance
500 spectrometer and was referenced against neat, external nitromethane
(coaxial capillary). The signals were not unequivocally assigned to the N
atoms.
15. The
spectrum was obtained on a Shimadzu QP 1000 instrument (EI, 70eV).
Sample Availability: Available from MDPI.
© 2006 MDPI. All rights reserved.