|
Molbank 2006, M520 |
Synthesis and Detailed Spectroscopic Characterization
of Two Novel N-(3-Acetyl-2-thienyl)acetamides
Gernot A. Eller* and Wolfgang Holzer
Department of Drug Synthesis, Faculty of
Life Sciences, University of Vienna, Althanstrasse 14, A-1090 Vienna, Austria
Phone: +43-1-4277-55634,
Email: [email protected]
* Author to whom correspondence should be addressed
Received: 1 September 2006 / Accepted: 20 October
2006 / Published: 1 December 2006
Keywords: N-acylations, 3-acetylthiophen-2-amine, NMR spectroscopy
Abstract:
The title compounds ¨C N-(3-acetyl-2-thienyl)-2-bromoacetamide and N-(3-acetyl-2-thienyl)-2-phthalimidoacetamide ¨C were synthesized in one step from 3-acetylthiophen-2-amine and the corresponding acetyl halogenides. Detailed spectroscopic data (1H NMR, 13C NMR, 15N NMR, MS, IR) for these compounds are presented.
Recently, we have investigated a modified Gewald reaction [1] for the preparation of 3-acetyl-2-aminothiophenes [2]. We here report the synthesis of two acetamides derived from 3-acetylthiophen-2-amine (1) (Scheme 1). These molecules are expected to be versatile intermediates for advanced investigations regarding the chemistry of 3-acetylthiophenes of type 1.
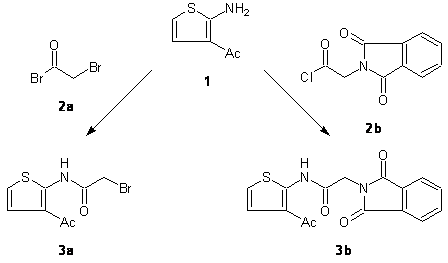
Scheme 1. Preparation of the title compounds 3a and 3b
N-(3-Acetyl-2-thienyl)-2-bromoacetamide
(3a):
Under
stirring at room temperature, to 4.23 g (30 mmol) thiophenamine 1 [2] in 70 mL of dry 1,4-dioxane were
added dropwise 6.06 g (30 mmol) of bromoacetyl bromide (2a) in 20 mL of 1,4-dioxane. After 3 h the reaction mixture was
poured into ice-cold H2O (ca. 300 mL), the resulting precipitate was
filtered off, washed with H2O, and dried under reduced pressure to
afford pure 3a (4.72 g, 60%) as a
beige powder. The compound slowly decomposes in DMSO- or MeOH-solution.
Melting point: 96¨C97ˇăC.
IR (KBr) [3]: 1660, 1640 cm¨C1.
1H NMR
(300 MHz, DMSO-d6) [4]: ¦Ä
(ppm) 12.18 (s,
1H NMR
(500 MHz, CDCl3) [5]: ¦Ä (ppm) 12.66 (s,
13C NMR (75 MHz, DMSO-d6) [4]: ¦Ä (ppm) 195.8 (COCH3), 164.6 (NCO, 2J(NCO,CH2) = 4.3 Hz, 2J(NCO,NH) = 4.3 Hz), 147.0 (C2), 125.2 (C4, 1J = 170.2 Hz, 2J(C4,H5) = 4.2 Hz), 121.8 (C3), 117.1 (C5, 1J = 189.3 Hz, 2J(C5,H4) = 6.0 Hz), 29.0 (CH2, 1J = 155.6 Hz), 28.8 (CH3, 1J = 127.7 Hz).
13C NMR (125 MHz, CDCl3) [5]: ¦Ä (ppm) 196.0 (COCH3, 2J(COCH3,CH3) = 5.9 Hz, 3J(CO,H4) = 0.9 Hz), 164.1 (NCO, 2J(NCO,CH2) = 4.5 Hz), 148.3 (C2, 2J(C2,NH) = 2.1 Hz, 3J(C2,H4) = 10.0 Hz, 3J(C2,H5) = 7.6 Hz), 124.4 (C4, 1J = 168.8 Hz, 2J(C4,H5) = 3.6 Hz), 122.0 (C3, 2J(C3,H4) = 5.8 Hz, 3J(C3,H5) = 9.1 Hz), 3J(C3,CH3) = 1.3 Hz), 116.9 (C5, 1J = 187.7 Hz, 2J(C5,H4) = 5.0 Hz), 28.7 (CH3, 1J = 127.9 Hz), 27.9 (CH2, 1J = 153.9 Hz).
15N NMR (50 MHz, CDCl3) [6]: ¦Ä (ppm) ¨C248.9 (NH).
MS (m/z, %) [7]: 263 (M+, 27), 261 (M+, 25), 141 (100), 126 (75), 43 (59).
Elemental Analysis: Calculated for C8H8BrNO2S (262.12) ∙ 0.1 H2O: C, 36.41%; H, 3.13%; N, 5.31%. Found: C, 36.15%; H, 2.92%; N, 5.03%.
N-(3-Acetyl-2-thienyl)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)acetamide (3b):
At room
temperature, to 2.12 g (15 mmol) of thiophenamine 1 [2] in 20 mL of dry 1,4-dioxane were added dropwise 3.35 g (15
mmol) of phthalimidoacetyl chloride (2b)
[8] in 20 mL of 1,4-dioxane. The reaction mixture was stirred overnight and
then poured into H2O (ca. 100 mL). Upon neutralization with solid
NaHCO3 a yellowish precipitate was formed which was filtered off,
washed with H2O, and dried under reduced pressure to afford pure 3b (4.33 g, 88%) as a yellowish powder.
Melting point: 208¨C212 ˇăC.
IR (KBr)
[3]: 1773, 1719, 1693, 1635 cm¨C1.
1H NMR (500 MHz, CDCl3)
[5]: ¦Ä (ppm) 12.25 (s, 1H, NH), 7.91 (m, 2H, Phth-H3,6), 7.76 (m, 2H,
Phth-H4,5), 7.17 (d, 3J(Th-H4,Th-H5)
= 5.8 Hz, 1H, Th-H4), 6.75 (d, 3J(Th-H5,Th-H4)
= 5.8 Hz, 1H, Th-H5), 4.65 (s, 2H, CH2), 2.49 (s, 3H, CH3).
13C NMR (125 MHz, CDCl3) [5]: ¦Ä (ppm) 196.1 (COCH3), 167.4 (Phth-CO), 164.1 (HNCO), 148.5 (Th-C2), 134.4 (Phth-C4,5), 131.9 (Phth-C1,2), 124.2 (Th-C4), 123.8 (Phth-C3,6), 121.5 (Th-C3), 116.6 (Th-C5), 40.7 (CH2), 28.6 (CH3).
15N NMR (50 MHz, CDCl3) [6]: ¦Ä (ppm) ¨C228.8 (NCH2), ¨C252.7 (NH).
MS (m/z, %) [7]: 328 (M+, 17), 168 (17), 160 (100), 141 (27).
Elemental Analysis: Calculated for C16H12N2O4S (328.34) ∙ 0.1 H2O: C, 58.21%; H, 3.72%; N, 8.49%. Found: C, 57.86%; H, 3.87%; N, 8.40%.
References
and Notes
1. Gewald,
K. Chem. Ber. 1965, 98, 3571¨C3577.
2. Eller,
G. A.; Holzer, W. Molecules 2006, 11, 371¨C376.
3. The spectrum was obtained on a Perkin-Elmer FTIR 1605 spectrophotometer.
4. The spectrum was obtained on a Varian UnityPlus 300 spectrometer (299.95 MHz for 1H, 75.43 MHz for 13C) at 28 ˇăC. The center of the solvent signal was used as an internal standard which was related to TMS with ¦Ä 2.49 ppm (1H NMR) and ¦Ä 39.5 ppm (13C NMR).
5. The spectrum was obtained on a Bruker Avance 500 spectrometer (500.13 MHz for 1H, 125.77 MHz for 13C) at 294 K. The center of the solvent signal was used as an internal standard which was related to TMS with ¦Ä 7.26 ppm (1H NMR) and ¦Ä 77.0 ppm (13C NMR).
6. The spectrum was obtained on a Bruker Avance 500 spectrometer (50.68 MHz for 15N) and was referenced against neat, external nitromethane (coaxial capillary).
7. The spectrum was obtained on a Shimadzu QP 1000 instrument (EI, 70eV).
8. Usifoh, C. O.; Lambert, D. M.; Wouters, J.;
Scriba, G. K. E. Arch. Pharm. (Weinheim,
Ger.) 2001, 334, 323¨C331.
© 2006 MDPI. All rights reserved.