http://www.chemistrymag.org/cji/2000/024019pe.htm |
Apr.24, 2000 Vol.2 No.4 P.19 Copyright |
Study on the stability of rhodium complex with uncoordinating donor and the property of catalysis
Jiang Hua, Diao Kaisheng#, Pan
Pinglai, Lu Kaijuan, Yuan Guoqing*
(Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100080, China; #Diao
Kaisheng, Guangxi Institute for Nationalities, Nanning, 530006)
Received
Feb. 1, 2000; Supported by the National Nature Science Foundation of China. (Grant No. 29974035)*Corresponding author
Abstract A new kind of rhodium
complexes with outstanding stability is reported here. On the basis of the evidence of its
structure and its data of IR and XPS, the existence of uncoordinating donor near center
rhodium atom has been found to be the radical reason for its high stability. An
intramolecular substitution reaction, which takes place between the center rhodium atom
and its near uncoordinating donor, prohibits rhodium complexes from decompostion due to
the loss of terminal carbonyls. On the contrary, a model complex without uncoordinating
donors exhibits lower catalytic stability. The result confirms that the existence of
uncoordinating donors greatly increases the stability of the complex in methanol
carbonylation reaction.
Keyword Rhodium complex, Stability, Methanol carbonylation
As catalyst in producing acetic acid
and acetic anhydride from methanol carbonylation, rhodium complexes are widely used
because they exhibit high catalytic activity under mild condition [1]. Their
high catalytic activity made them the most successful catalysts in carbonylation reaction.
However, they are usually unstable when heated without the efficient protection of CO.
They tend to change into RhI3 and lose their catalytic activity. This make it
very difficult to protect catalyst in the process of separating products from catalyst[2].
We had reported many copolymer rhodium complexes, such as PVMRh, YDRh
and PMARh. They all have high catalytic activity as well as high stability[3,4,5].
These copolymer rhodium complexes are the first kind of catalysts that both have high
catalytic activity and good stability, which threw a light into the situation that the
catalyst has high activity but has no good stability in carbonylation reaction. In order
to design new catalysts with excellent capability, it is very important to find the reason
for their excellent performance. Some complexes with high stability had been studied here
and the results show that the existence of uncoordinating donors near center rhodium atoms
is the radical reason for their high stability. Here, we systematically studied a kind of
square planar rhodium complex with uncoordinating donor near the center rhodium. Following
the loss of terminal carbonyl attributed to lack of the efficient protection of CO, the
uncoordinating donors in ligand will immediately coordinate with center rhodium atoms and
replace the lost terminal carbonyl. And it helps to prevent the complex from decomposition
due to the loss of the terminal carbonyl. it should be noted that the intramolecular
substitution reaction is reversible. The above complex will revert to its original
structure under an atmosphere of CO. This intramolecular substitution reaction largely
enhances the stability of the complex in carbonylation from methanol to acetic acid.
Besides, further evidence for the influence on its catalytic stability have been studied
by comparing its model compound. The resultant evidence conforms that the above conclusion
is sound and well granted.
1 EXPERIMENTAL
1.1 Preparation of ligands and complexes
1.1.1 Preparation of ligand PM and MM The intermixture of
Mercaptoacetic acid(M) and 1,3-Propandiol(P), in the ratio M:P=7:3(mol), was refluxed at
100°C for 5h under the catalytic action of sulfuric acid to
give a clear yellow solution. After cooling the reacted solution to room temperature, an
aqueous solution of sodium hydrogen carbonate was added dropwise to it. The lower organic
layer was separated and washed several times with the aqueous solution of sodium hydrogen
carbonate and water. After drying the liquid over anhydrous magnesium sulfate, a colorless
liquid was obtained. And so does the preparation of ligand MM, which derives from
Mercaptoacetic acid (M) and Methanol (M).
1.1.2 Preparation of complex PMRh and MMRh A tetrahydrofuran solution of
[Rh(CO)2Cl]2 was added dropwise to a tetrahydrofuran solution
containing excessive PM under stirring to give a mixture solution. A tetrahydrofuran
solution of excess NaBPh4 was then added to the above mixture solution to give
a yellow complex PMRh. The product was filtered, washed with diethyl ether and dried. And
so does the preparation of complex MMRh.
1.2 Characteristic of the complexes
The XPS measurement of the complexes and ligands were conducted on a Kratos ES 300
X-ray photoelectron spectroscopy, using Mg Ka1,2 X-radiation at 1 254 eV with the vacuum of 2.7*10-7
Pa. Its IR spectrum was recorded on a VR-10 spectrophotometer.
1.3 Carbonylation of methanol to acetic acid
Methanol, methyl iodide and prepared catalysts were added to an autoclave in a given
proportion. The products were analyzed by GC-103 gas chromatography with a column length
of 2 m at 150°C , and H2 with the velocity of 40
ml/min was used as the carrier.
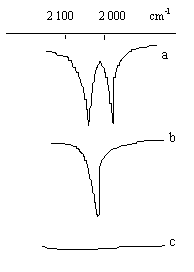 |
Table 1 Data of the IR spectroscopy nCO(cm-1)
for the terminal carbonyl of the complex PMRh
|
|||||||||||
| Fig.1 Characteristic absorption peaks of terminal carbonyl |
2 RESULTS AND DISCUSSION
2.1 Intramolecular substitution reaction and studies on IR and XPS spectrum
The ligand PM contains four donors, including two sulfur atoms and two oxygen. When the
ligand PM reacts with [Rh(CO)2Cl]2 in tetrahydrofuran solution, one
of the sulfur atom coordinates with rhodium atom to form a strong S![]() Rh bond. With the
progressive addition of NaBPh4 to the solution, the Cl-Rh bond was broken and
the oxygen atom near the center rhodium atom coordinated with rhodium to form a weaker O
Rh bond. With the
progressive addition of NaBPh4 to the solution, the Cl-Rh bond was broken and
the oxygen atom near the center rhodium atom coordinated with rhodium to form a weaker O![]() Rh bond and then to give
the complex remaining two uncoordinated donors. Table 1 shows the data of IR spectroscopy
reflected the terminal carbonyl in complex PMRh. When the complex PMRh (whose IR spectrum
shows in Fig.1 a) was placed at room temperature over 24 hours, the double peaks, which
reflects the characteristic absorption of terminal carbonyl groups, changed into single
peak (Fig.1 b). It indicates that one of the two terminal carbonyl had dropped off[6,7].
Furthermore, the single peak also disappeared when the complex was heated at 100°C for 1.5hr (Fig.1 c) and no peak reflected terminal carbonyl can be
seen up to now. It is surprising that the double peaks would recover in its IR spectrum
(similar to Fig.1 a) if the treated complex was heated at 100°C for
2hr under an atmosphere of CO (above changes were showed in Fig.1). According to above
phenomena, we propose a reversible intramolecular substitution reaction as folllow:
Rh bond and then to give
the complex remaining two uncoordinated donors. Table 1 shows the data of IR spectroscopy
reflected the terminal carbonyl in complex PMRh. When the complex PMRh (whose IR spectrum
shows in Fig.1 a) was placed at room temperature over 24 hours, the double peaks, which
reflects the characteristic absorption of terminal carbonyl groups, changed into single
peak (Fig.1 b). It indicates that one of the two terminal carbonyl had dropped off[6,7].
Furthermore, the single peak also disappeared when the complex was heated at 100°C for 1.5hr (Fig.1 c) and no peak reflected terminal carbonyl can be
seen up to now. It is surprising that the double peaks would recover in its IR spectrum
(similar to Fig.1 a) if the treated complex was heated at 100°C for
2hr under an atmosphere of CO (above changes were showed in Fig.1). According to above
phenomena, we propose a reversible intramolecular substitution reaction as folllow:
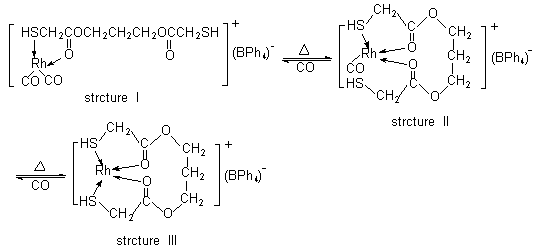 |
Scheme 1 Intramolecular substitution reaction |
When the complex
PMRh (structure I) was heated without efficient protection of CO, one of the two terminal
carbonyls dropped off. In the meantime, the neighboring uncoordinating oxygen donor
immediately replaces its place to form a O![]() Rh bond. So we got complex of structure II. Moreover,
the uncoordinated sulfur donor will replace the remaining terminal carbonyl to form
quadridentate complex of structure III if the complex was heated further. And it should be
noted that this substitution reaction is reversible. The complex of structure III will
revert to its original complex of structure I Under an atmosphere of CO. That is the
reason why the IR spectrum changes in the complex PMRh.
Rh bond. So we got complex of structure II. Moreover,
the uncoordinated sulfur donor will replace the remaining terminal carbonyl to form
quadridentate complex of structure III if the complex was heated further. And it should be
noted that this substitution reaction is reversible. The complex of structure III will
revert to its original complex of structure I Under an atmosphere of CO. That is the
reason why the IR spectrum changes in the complex PMRh.
From the XPS of ligand PM and complex PMRh in three different
structures, we can draw the data of the binding energy of S2P , O1S
and Rh 3d5/2 . As shown in Table 2, S2P binding energy of
ligand is 162.0 eV. After some sulfur atoms coordinating with the rhodium atom in
structure I, part of the S2P binding energy of complex PMRh increased to 163.3
eV but that of the uncoordinating sulfur remained constant. As for structure II, the
uncoordinating carbonyl oxygen atom replaced the lost terminal carbonyl to form tridentate
complex. Because the coordinating status of sulfur did not change, its XPS spectrum did
not change, too. With the loss of the second terminal carbonyl, the uncoordinating sulfur
donor immediately coordinated with rhodium atom to form stable quadridentate complex of
structure III. As a result the S2P binding energy all increased to 163.4 eV due
to no uncoordinated sulfur atoms. the same was the case with the changes of O1S
binding energy.
Table 2 Binding energies of S2P , O1S and Rh3d5/2 of complexes and ligand
S2P binding energy Eb(eV) |
O1S binding energy Eb(eV) |
Rh3d5/2 binding energy Eb(eV) |
|||
[Rh(CO)2Cl]2 |
533.2 |
311.2 |
|||
ligand PM |
162.0 |
533.1 |
|||
structure I |
162.1* |
163.3** |
533.0* |
534.6** |
310.1 |
structure II |
162.1* |
163.3** |
533.1* |
534.5** |
309.5 |
structure III |
163.4** |
533.1* |
534.5** |
308.5 |
|
Note: * present uncoordinated atom of complex; ** Present coordinated atom of complex.
From Table 2 we can also see that the changes of
Rh3d5/2 binding energy is apparent. It decreased to 310.1 eV in structure I,
comparing with that of 311.2 eV in [Rh(CO) 2Cl]2.This indicates that
rhodium accept the negative charge from sulfur and oxygen donors during the formation of
complex PMRh. With the complex changing from structure I to structure II, the binding
energy of Rh3d5/2 decreased to 309.5 eV. And it further decreased to 308.5 eV
when the structure changed from II to III.The changes of the binding energy conform that
the above intramolecular substitution reaction is reasonable.
2.2 Catalytic activity
The above intramolecular substitution reaction greatly enhances the stability of the
complex. However, the reaction took place in a complicated system when the complex was
used as catalyst for carbonylation of methanol to acetic acid. In order to test its
catalytic activity in methanol carbonylation, we designed a synthesized model compound
MMRh, whose structure is similar to that of complex PMRh but has no uncoordinating donors. The structure of complex PMRh and of its model
compound MMRh are showed in Scheme 2.
Experiments of catalytic carbonylation of methanol were conducted to
test their activity. As shown in Fig. 2, methyl acetate is the main product at the
beginning of the reaction. With time going on, methyl acetate reaches the culmination and
begins to decrease. At the same time, acetic acid becomes the main product. This trend is
consistent with our former work.
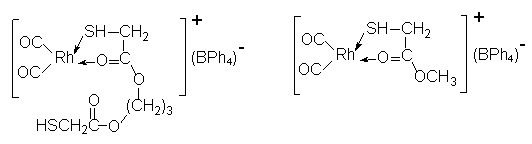 |
| PMRh MMRh Scheme 2 Structure of the complexes |
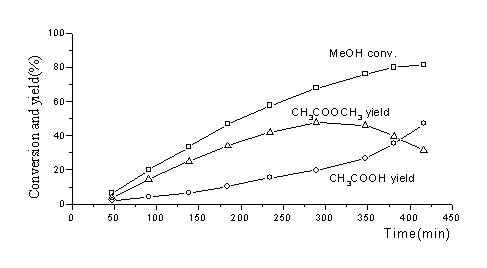 |
Fig. 2 Conversion and yield of
methanol carbonylation in catalyst PMRh |
Because the structure of PMRh is similar to that of MMRh, they both should have the same catalytic activity in methanol carbonylation reaction. Respectively catalyzed by PMRh and MMRh, the comparing experiments were carried out in the same conditions. As shown in Table 3, both the conversion of methanol and the yield of acetic acid and methyl acetate are nearly the same. It indicates that the two exhibit a close catalytic activity.
Table 3 Conversion of methanol and yield of acetic acid and methyl acetate
| complex | conversion of methanol (%) |
yield of acetic acid (%) |
yield of methyl acetate (%) |
PMRh |
82.0 |
31.6 |
50.4 |
MMRh |
79.1 |
34.0 |
45.1 |
Reaction condition: the same above
2.3 Catalytic stability
However, their catalytic stability are greatly different. Fig. 3 is the relationship
between the rate of CO absorption and time. In the curves, aerating air into the autoclave
to destroy the catalyst at the same time (A and C points), Curves AB and CD present the
latent period to reactivate the catalyst. As we can see, the latent period of catalyst
PMRh is much shorter than that of MMRh. And the rate of CO absorption in PMRh has no
change after reactivated. The resultant evidence conforms that PMRh maintains its
catalytic activity. On the contrary, the rate of CO absorption in MMRh is much lower than
that of it prior to aerate air. Because the catalyst MMRh has been destroyed by air due to
lack of the efficient protection of CO. This experiment strongly confirms that the
intramolecular substitution reaction is the radical reason for its high stability.
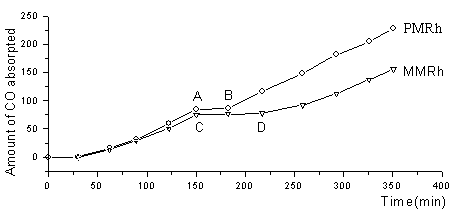 |
| Fig. 3 Relationship between the amount of CO absorpted and time Reaction condition : the same above |
On the basis of the evidence of IR, XPS and catalytic comparison experiment, we come to the conclusion that the protection of uncoordinating donor near center rhodium atom is the radical reason for its high stability. If complex PMRh is heated without efficient protection of CO, the uncoordinating donors can immediately replace the losing terminal carbonyl to form a stable quadridentate complex. And it helps to prevent the catalyst from decomposing. That is the reason why complex PMRh has high stability. The intramolecular substitution reaction makes a way to alleviate the problem about the catalyst with high activity but without high stability.
REFERENCES
[1] Fordye W A, Crosby G A. Inorg. Chem., 1982, 21: 1023.
[2] Jareel M S, Gates B C. J. of Cat., 1975, 40: 225.
[3] Yuan G Q. Acta Chimica Sinica, 1983, (1): 86.
[4] Yuan G Q, Liu Z Y, Pan P L et al. Acta Polymerica Sinica, 1993, (3): 375.
[5] Pan P L, Liu Z Y, Yuan G Q et al. Chemistry (Huaxue Tongbao), 1996, 9 (1): 25.
[6] Yuan G Q, Wang D H, Liu S H et al. (Kexue Tontbao), 1984, 29 (8): 1053.
[7] Liu Z Y, Pan P L, Huang M K, Yuan G Q. Journal of Functional Polymers, 1994, 7
(2): 159.