http://www.chemistrymag.org/cji/2001/032006pe.htm |
Feb. 1, 2001 Vol.3 No.2 P.6 Copyright |
Photocatalytic decomposition of P-chloroaniline in water over immobilized TiO2
Leng
Wenhua, Tong Shaoping, Cheng Shao, an, Zhang Jianqing, Cao Chunan
(Department of Chemistry, Yuquan Campust, Zhejiang University, Hangzhou,
310027, China)
Abstract In the present study, the
photocatalytic decomposition of p-chloroaniline (PCA) in water was explored using titanium
dioxide as catalysts immobilized on porous nickel with binder. The results showed that PCA
can be destroyed effectively by this method in the presence of oxygen. The degradation
rate is strongly dependent on the initial PCA concentration, pH and oxygen concentration.
The initial quantum yield is 4.54% for PCA 8.65×10-5 mol·dm-3.
Total mineralization required much longer illumination time than the disappearance of PCA.
Without oxygen, the photocatalytic reaction could be performed via imposing external
anodic bias. The degradation kinetic of PCA can be described by Langmuir-Hinshelwood
equation, and the main immediate products of PCA were aniline, 4-chloronitroaniline,
azobenzene, 4-4' - dichloroazobenzene and nitrobenzene, which
were mineralized to NH4+、Cl-、NO3- and CO2.
Keywords Photocatalytic decomposition; Titanium dioxide; Immobilization; P-chloroaniline
Titanium dioxide photocatalysis has been
demonstrated a promising method [1, 2] for the pollutant treatment mainly due
to its capability of complete mineralizing or at least partly destroying a variety of
organic pollutants. However, much attention in this area has focused on the use of slurry
system, about which the need to separate the spent fine catalysts particles and to keep
the semiconductor suspended is usually necessary. Recently, some researches have tried to
minimize these problems by immobilizing TiO2 on various materials for
photocatalytic reaction, such as glass [3, 4], sea sand and gel [5],
conductive glass [6, 7], titanium [8-10] and stainless steel
[11], etc. Most of those methods used for the adhesion of photocatalysts on supports
need heating. This may bring some disadvantages: (a) some porous structure gets lost, thus
inherently a decrease in the surface area available for reaction and an impedance to the
transportation of solutes arises, consequently the efficiency of photocatalytic reaction
will decrease; (b) it requires to control strictly the heating temperature to preserve
photocatalytic activity.
We have explored a new simple fixation technique of TiO2
bound by binder on a porous nickel support for the photocatalytic degradation of
sulfosalicylic acid that is strongly adsorbed to TiO2 and nickel, the results
is satisfactory [12]. Porous nickel was chosen as support in view of its
following characteristics: (a) high porosity suitable for catalyst fixation and the
transportation of solutes; (b) good conductivity capable of imposing an external potential
bias on it to improve the photogenerating carries separation, and (c) low cost and wide
application in industry, for example as current collector in batteries.
PCA is commonly produced as by-product of some petroleum and chemical
industries, is highly toxic. The studies of its degradation have been investigated by many
methods such as hydrogen peroxide [13] and electro-oxidation [14] and
the photocatalytic degradation in slurry system [15]. In this study, we report
its photocatalytic degradation over immobilized TiO2. The effect of the initial
PCA concentration, pH, oxygen concentration, temperature and external potential on the
photocatalytic reaction rate was investigated. The quantum yield of degradation reaction
and the formation of Cl-, NO3-, NH4+
ions and CO2 were also measured. The degradation kinetic and mechanism were
also discussed.
1 EXPERIMENTAL
1.1 Materials
TiO2 (C. P. 99.9%, Shanghai) was used as received. Polyvinyl alcohol (PVA 124)
used as binder was purchased from Japan. Porous nickel sheet (thickness about 0.2 cm) was
used as support. PCA were analytical grade and used without further purification; KNO3
were governmental reagent; H2O2 were 29 wt. %. The other
chemicals and solvents were reagent grade and used without further purification. Deionized
and double distilled water was used throughout the work.
1.2 Immobilization of catalysts
The procedures of fixation of TiO2 were similar to that described in ref.12, i.
e., TiO2 was mixed with 3-wt. % PVA at the ratio of 3 to 2. It was coated on
one side of a 15×16 cm porous nickel sheet. The coated sheet (denoted hereafter as TiO2/Ni)
was dried at 60°C for 4 hour and packed to thinner sheet in the thickness of
0.09 cm. Then it was rinsed with saturated Na2CO3 to remove grease
and/or photooxidation products and washed with water to release the loosely bound
particles and dried by heating at 60°C prior to use or reused. In
photoelectrochemical experiments, the nickel double sides covered with TiO2 was
fixed on the outer surface of the borosillicate glass tube as working photoelectrode
(Blank porous nickel as counter electrode, a double-junction saturated calomel electrode
as reference, 0.001 mol dm-3 NaClO4 as support electrolyte). The
bare nickel in same thickness was used as blank tests. The difference in weight between
the coated and bare films gave the TiO2 weight as listed per gram nickel in the
following experiments. We may not obtain the same weight of TiO2, surface area,
and etc on different TiO2/Ni sheet, so the results can not compare with each
other in the text. But for comparison the contrast experiments were conducted on the same
TiO2/Ni sheet.
1.3 Characterization of TiO2/Ni
X-ray diffraction analysis (XRD) was carried out with CuKα radiation on a Rigaku D/max 3B
diffractometer (working at 45 kV, 40 mA, at a speed of 10 deg min-1 and a step
0.02 deg. ).
The scanning electron microscopy (SEM) was carried out on a low vacuum
SEM Hitachi (S-570) with an energy dispersive x-ray (EDX) analysis attachment.
1.4 Photoreactor and light source
Photoreactor was an annulus reactor with a F6.5 cm cylindrical borosillicate glass inner tube (Thickness about
3 mm) around which the TiO2/Ni was wrapped. The light source (4×6 W UV lamp,
Emax=365nm) was fixed on the central axis of the tube. The number of the incident light
(300-400 nm) inside the photoreactor in TiO2/Ni free solutions measured
employing potassium ferrioxalate actinometry [16] was 1.4×10-7 mol·s-1.
1.5 procedure and analyses
The solution (180 cm3) was placed in the annular region (width about 0.5 cm)
between the tube and the double-walled glass outer jacket (cooling at 25+1°C),
magnetically stirred and kept purged at the side of photoreactor with air at a rate of 16
dm-3 h-1 and initial pH 7.0 unless otherwise stated. The PCA
concentration in the solution was estimated colorimetrically by the modified method [17]
(coupling time was 2 hour). The degradation products NO3- and
NH4+ were estimated by indophenol blue [18] and phenate
method [18], respectively. Chloride ion concentrations were measured with a
chloride ion-selective electrode and double-junction reference electrode by withdrawing
20cm3 solution from the reactor and returning after measurement. HClO4
and/or NaOH adjusted the pH of solution before reaction.
Organic solutions were analyzed by gas chromatography/mass spectrometry
(GC/MS, HP 6890/5973) with a HP5 column (30 m×0.25 mm). The oven was programmed as
follows: isothermal at 45°C for 2 min, from 45°C -250°C
at 6°C min-1, and isothermal at 250°C for 10 min. In
the electron impact experiments, the electron energy was set at 70 eV and the electron
current was set at 1 mA.
2 RESULTS AND DISCUSSION
2.1 Characterization of TiO2/Ni
XRD analysis showed that TiO2 powder was almost anatase phase and the average
particle size was about 114 nm calculated by Scherrer formular. The specific surface of
TiO2 was 14.98 m2 g-1 (measured on Omnisorp 100 CX,
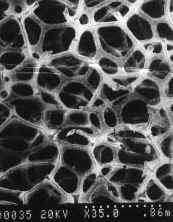
Coulter, USA). The texture and morphology of the porous nickel and TiO2/Ni can
be observed in Fig. 1. SEM analysis showed that the nickel substrate was meshy and high
porous (95%), the diameter of pores was in the range of from 5mm to 50m m. Thus it is
favorable for the fixation of TiO2. TiO2 was dispersed in the top of
the pores of the nickel sheet but there remained many pores inside of the TiO2/Ni,
The diameter of its pore was less than 10 mm.
2.2 Influence of the initial concentration
It is found that a negligible decrease in the concentration of PCA for 8h was observed
both in the presence of TiO2 without UV light irradiation (3% degradation) and
without TiO2 but with illumination (8% degradation). However, PCA can be
degraded quickly over illuminated TiO2 (as shown in Fig. 2), it resulted
obviously from photocatalytic oxidation. The rate increase with increasing PCA
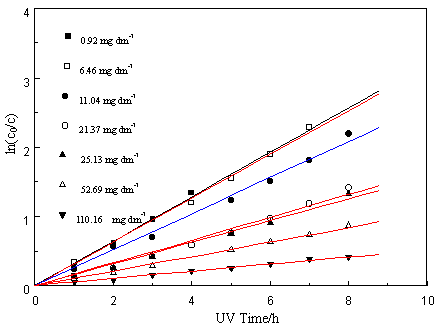
concentration as indicated in Fig. 2b and the photocatalytic degradation reaction follows
a quasi-first-order in the range of a studied PCA concentration, which is concluded from
the straight-line relationship of ln (co/c) vs. irradiation time as
shown in Fig. 2a. The slope of the plot gives the apparent first-order rate constant kapp
as listed in Table 1. It is clear that the apparent rate constants decrease with
increasing PCA concentration. This can be explained by assuming that the photoproducts
were competition for the surface site of TiO2 with PCA. Table 1 also listed the
half-lives and quantum yields of the photodegradation of PCA as a function of initial
concentration.

(a)
(b)
Fig.1 SEM micrographs for (a) the porous nickel and (b) TiO2/Ni
 |
(a) |
 |
(b) |
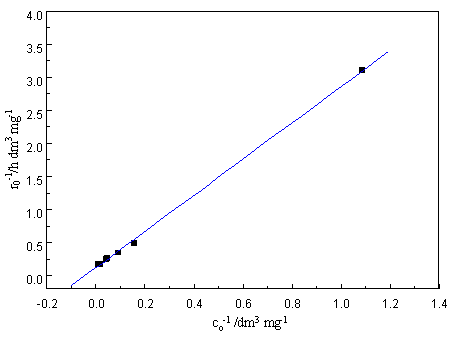
Fig.2 (a) Pseudo-first-order kinetics for PCAs in photocatalytic degradation (b) Linearized reciprocal kinetic plot for the photocatalytic degradation of PCA(Experimental conditions: TiO2, 1.48g/g (Ni)
Table 1. Apparent first-order rate constants kapp, half life t1/2, t1/2', quantum yield j and linear coefficient r2 for the degradation of PCA at different initial concentrations (Experimental conditions see Fig. 2)
| Initiate concentration (mg·dm-3) |
kapp. |
t1/2 |
t1/2' |
j |
r2 |
0.92 |
34.973 |
1.98 |
1.96 |
0.5 |
0.981 |
6.46 |
32.049 |
2.16 |
2.28 |
3.21 |
0.997 |
11.04 |
26.409 |
2.63 |
2.54 |
4.54 |
0.993 |
21.37 |
17.768 |
3.90 |
3.14 |
5.90 |
0.990 |
25.13 |
16.509 |
4.20 |
3.36 |
6.45 |
0.994 |
52.69 |
10.843 |
6.39 |
4.96 |
8.88 |
0.997 |
110.16 |
5.326 |
13.01 |
8.29 |
9.13 |
0.995 |
2.3 Influence of the initial pH
Fig. 3 shows the dependence of degradation of PCA on the initial pH solution. The
increasing r0 with increasing pH can be attributed to the increase number of OH-
ions at the surface of TiO2 ( OH·
radical can be formed by trapping photoproduced holes). Also the dissociation of PCA
probably changes its reactivity. Claire [19] found that OH·
radicals were the sole oxidant under the condition of pH 11 and its role was larger in
this case than in neutral and acid medium. So r0 increases rapidly with
increasing pH higher than 11. But at the lowest pH (to avoid Ni dissolving, lower than pH
4 not investigated) the rate is influenced at least by two factors. One in the case it is
lack of OH- ions, thus produce less OH· radicals,
the rate decreases; another it can provide more H+ that contributes the
formation of HO2- [20], thus the rate
increase. In the experiments the rate increases slightly that may be attribute to the
dominant role of the latter. In general, the results indicate the efficiency of the
process is not much affected over a wide moderate range of pH, which is quite satisfactory
in view of applications.
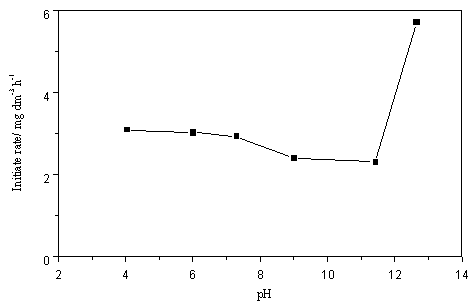 |
Fig.3
Effect of the initial pH on the initial rate of PCA in photocatalytic degradation |
2.4 Influence of the oxygen
The influence of oxygen on the degradation of PCA is shown in Table 2. It is obvious that
the oxidation rate of PCA increases with increasing oxygen concentration. This has well
been demonstrated early by many studies [1, 2]. Oxygen is often as an electron
acceptor without which the photocarriers will be totally combined and it may be also an
oxidant in the further process of the hydroxyl of organic substance. In addition,
adsorptive oxygen at the surface of particles is another source of OH·
radical via a series of chemical process [20].
It also can be observed from the Table 2 that the formation rate of
chloride ions is larger than the disappearance of PCA both in the case of air and oxygen
purge. Similar results were obtained in the degradation of chloramben [21].
They explained that hydroxyl radicals usually attack the aromatic substrates to form the
corresponding OH· adducts still adsorbed onto the oxide
particles where they can interact with the trapped electrons to release chloride.
2.5 Influence of the temperature
Table 3 lists the apparent rate constant as a function of temperature for PCA degradation
8h. The Arrhenius equation allowed a calculation of the activation energy of the PCA
degradation of 6.12 kJ·mol-1 (r=0.993), which is less than the value of 10 kJ
mol-1 obtained for phenol [20], 11 kJ mol-1 for
salicyclic acid [3], 13 kJ mol-1 for oxalic [24]. This
indicated that PCA is easily degraded.
Table 2. Effect of oxygen content on the half life of PCA (t1/2, PCA) and chloride removal (t1/2, cl) for the PCA photocatalytic degradation (C0, PCA =112.41mg dm-3; TiO2= 1.98 g/g (Ni))
| O2 content (v/v, %) | t1/2, PCA /h |
t1/2, cl /h |
20 |
11.02 |
3.72 |
50 |
7.96 |
- |
100 |
6.57 |
2.37 |
Table 3. Apparent first-order rate constants kapp, as a function of temperature (T) for the PCA degradation (Experimental conditions: C0, 11.47 mg dm-3; TiO2=1.93 g/g (Ni); UV 8h)
| T/K | kapp./(×102 h-1) |
298 |
13.316 |
305 |
13.664 |
313 |
14.791 |
333 |
17.096 |
2.6 Formation of nitrate ions and ammonium
ions
Photoproducts may be more toxic than the solute itself, so totally destroying the
pollutants is necessary. For evaluate what the extent of PCA mineralization performed,
photocatalytic degradation was conducted and the results are shown in Fig.4. It shows that
the PCA was completely disappeared when illuminated for 10 h, while only 51.0% and 20.0%
of NH4+ and NO3- was formed respectively. The
formation of NH4+ ions was rather quick in initial stage and then
became slow after reaction for 7h and 30min, while the formation of NO3-
increased slowly all the time with increasing illumination time. These indicated that the
remaining of nitrogen exists in others compounds and the oxidation of nitrogen to NO3-
may occur via the intermediate formation of NH4+ [15].
Similar results were found in photocatalytic degradation of PCA in slurry system when the
illumination time is not longer enough [15]. Fig.4 also shows that the
degradation of COD is a more slow process. All these indicated total mineralization
requires a much longer illumination time than the disappearance of PCA in the experiment.
It may be attributed to that the photoproducts were difficult to degrade and the light
intensity was rather poor.
2.7 Quantum yield
The quantum yield is an important parameter for photocatalytic process in practical
application and it can be defined as the ratio of the number of molecules of PCA reacting
to the number of photons supplied. Assuming that all the incident light was absorbed by
the titanium dioxide, under PCA 8.65×10-4 mol·dm-3 (other
experimental conditions see Fig. 2), the initial rate of PCA was found to be 3.81×10-7
mol·dm-3 min-1. Therefore the initial quantum yield for the
degradation of PCA is 4.54%.
2.8 Influence of the external potential bias
The photocatalytic reaction cannot proceed in the presence of nitrogen purge. For this
case, a test of photocatalytic degradation of PCA by applying an external anodic bias was
conducted and the results are shown in Fig. 5. It was found whether in the presence of UV
and external potential or with external potential alone, the reactions followed a
quasi-first-order expression. But the apparent first-order rate constant kapp
of the former is larger than that of the latter. For example, by exerting anodic 700 mV
the apparent quantum yield was 13.75% (not including the part of direct
electro-oxidation). The trend of between apparent first-order rate constants kapp
and pH under potential bias 600 mV as shown in Table 4 is consistent with that of no
potential bias (see Fig. 3). These indicate that the anodic bias can be efficiently
improved photocatalytic reaction. The reason is that bias potential applied to the
photocatalyst film had separated the photogenerated carriers thereby improving the quantum
yield [6, 7].
So far we can see that this approach has the advantage of not only the
ease of treating the photocatalyst after use but also capable of applying a bias potential
to improve the efficiency of PCA photodegradation with nitrogen on TiO2/Ni. By
this to study the photoelectrochemistry behavior of organic substrates in details is under
way in our laboratory.
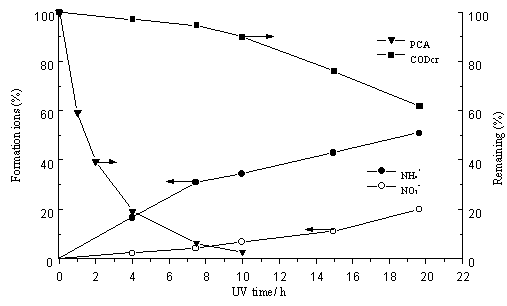 |
Fig.4 Ions formation, CODcr
and PCA degradation (Experimental conditions: C0, 22.70 mg dm-3; pH 8.70; TiO2= 2.05 g/g (Ni); oxygen purge) |
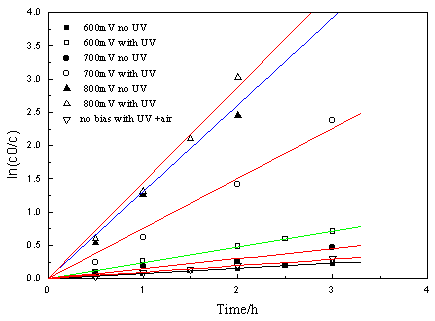 |
Fig.5 Effect of applied
potential (Vs SCE) on the photocatalytic degradation of PCA (Experimental conditions: C0, 9.80 mg dm-3; TiO2=3.95 g/g (Ni); N2 purge) |
Table 4 Apparent first-order rate constants kapp
and linear coefficient r for the PCA degradation 3h
at different pH with potential bias 600 mV (N2 purge, other experimental
conditions see Fig. 8)
| pH | kapp./(×102 h-1) |
r |
4.0 |
27.305 |
0.998 |
7.0 |
23.751 |
0.998 |
9.8 |
48.403 |
0.996 |
12.3 |
104.028 |
0.994 |
2.9. Kinetic and mechanism aspects
Langmuir-Hinshelwood (L-H) equation is often used to express the heterogeneous
photocatalytic reaction [1-3, 12, 25]. For the conditions in our experiments,
then it is given by
r =![]() (
(![]() +
+![]() ) (1)
) (1)
where r (mg dm-3 h-1)is
the reaction rate for the oxidation of PCA, c(mg
dm-3 )is the concentration of PCA, kr(mg dm-3 h-1)is the specific reaction rate constant, ka (dm3
mg-1) is the equilibrium adsorption constant of PCA, cw (mg dm-3
) is the concentration of solvent, kw (dm3 mg-1) is
the equilibrium adsorption constant of solvent, ci(mg dm-3)is the
concentration of products, ki (dm3 mg-1) is the
equilibrium adsorption constant of products and t (h) is the reaction time.
By integrating Eqn. (1), we get:
t =![]() (2)
(2)
If c0 is small, then the adsorption of
its product is negligible compare to reactant, i.e., ![]() . By substituting c=c0 into Eqn.(1)and (2), Eqn.(3)and
(4) can be obtained, respectively.
. By substituting c=c0 into Eqn.(1)and (2), Eqn.(3)and
(4) can be obtained, respectively.
![]() (
(
As above mentioned, c0
is small, so the second item in equation (4) can be neglected; for common system, cw
is a constant and it is much more larger than c, thus Eqn. (4) can be
written into: ![]() (5)
(5)
When it was plotted by ![]() and
and ![]() from equation(3)and
(5), straight line could be obtained, respectively. In addition, while by substituting
c0 c =c0/2 into equation (4), we
can get:
from equation(3)and
(5), straight line could be obtained, respectively. In addition, while by substituting
c0 c =c0/2 into equation (4), we
can get:
![]() (6)
(6)
Straight line can also be obtained when it was plotted by ![]() vs co from Eqn.(6).
vs co from Eqn.(6).
For our results as indicated in Fig.2a, the straight-line relationship
of ln (c0/c) versus irradiation time were observed. The plot of the reciprocal
initial rate r0-1 as a function of the reciprocal initial
concentration C0 yields a straight line (see Fig.2b). The linear transform of
this expression yields kr=8.622 mg dm-3 h-1 and ka=4.225×10-2
dm3 mg-1(r =0.9996). By substituting them into Eqn. (6), the estimated half-lives ![]() are obtained and listed in Table 1. The
dependence of
are obtained and listed in Table 1. The
dependence of ![]() on the initial
concentration of PCAs is also a straight line:
on the initial
concentration of PCAs is also a straight line: ![]() , (r2=0.995). So the photocatalytic oxidation of PCA in the
system can be well expressed by L-H model.
, (r2=0.995). So the photocatalytic oxidation of PCA in the
system can be well expressed by L-H model.
It has been proved [1, 2, 20] that photogenerated holes
oxidize water or adsorbed OH- at the surface of semiconductor to hydroxyl
radicals. These highly reactive radicals can then be used to mineralize or at least
partially degrade most organic pollutants. At the same time the holes can also react with
PCA directly to produce PCA cation radical. Due to the complexity of the radical-induced
reactions occurring in photocatalytic process, it is difficult to indicate an exhaustive
reaction scheme explaining the formation of all detected intermediates. However, a
relatively low number of rather abundant aromatic transients have been recognized during
the photocatalytic reaction, so that a tentative and simple scheme can be drawn, with
considering into the usual transformation processes of other aromatics. We have obtained 5
intermediates during the initial photocatalytic degradation of PCA by GC/MS as listed in
Table 5. Azobenzene (4- 4'-dichloroazobenzene) was formed from the coupling of neutral
free radical produced through the deprotonation of the aniline (PCA) cation radical which
was probably generated by reacting with hole[14]. Generation of NH4+
is considered to be produced by the hydrolytic degradation of imino intermediates leading
to the corresponding benzoquinone [14]. But we did not detect this compound by
GC/MS. Brillas et al. [13] also did not find it detected by GC/MS and HPLC
during the electrochemical oxidation of PCA. They did not think that this compound could
be ruled out as intermediate due to its rapidly decomposed by different paths. Hydroxyl
radicals attack the aromatic compound should be an important step to produce
hydroxyaromatic intermediates in photocatalytic reaction, for example, hydrohydroquinone
is the main intermediate product detected for phenol [20] and hydroxyaniline
for aromatic amines [15, 25]. However, they are usually unstable and difficult
to identify during the initial steps of the treatment.
So based on the above GC/MS results
we preliminary proposed the reaction scheme as shown in Fig. 6.
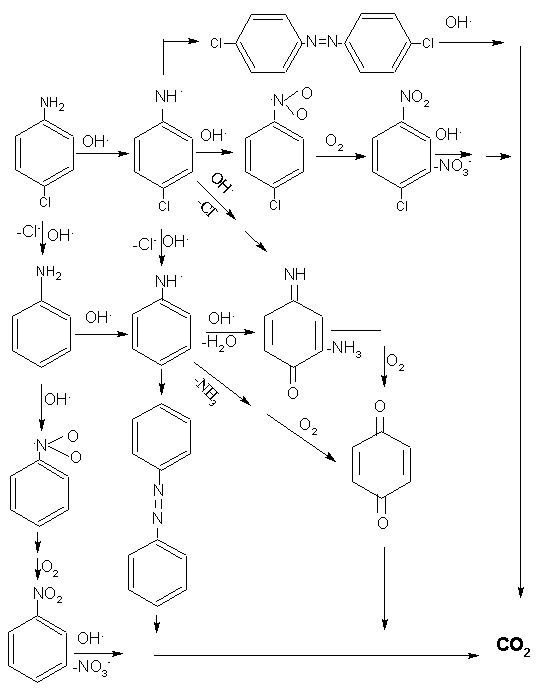 |
| Fig. 6 Supposed photocatalytic pathway of PCA |
Table 5 Identification of
photocatalytic immediates of PCA
(PCA 50 mg dm-3; pH12.5; oxygen purging; UV 1h)
| Compound | Molecular weight |
Main fragment (m/e) |
Aniline |
93 |
93,66,56 |
Nitrobenzene |
123 |
77,65,51 |
4-chloronitrobenzene |
157 |
75,111,50 |
azobenzene |
182 |
77,51,105 |
4-4'- dichloroazobenzene |
250 |
250,111,83 |
2.10 Further remarks about the TiO2/Ni
The coming off of powdered TiO2 from the substrate was not observed during the
process of photocatalytic degradation. Ni2+ concentration in solution measured
by atomic absorption was less than 1 mg L-1 when pH higher than 7.0 but less
than 70mg L-1 at pH 1.8 due to the dissolving for 7 days of the porous nickel.
This is because of the inhibition of TiO2 on bare nickel. Contrast tests were
conducted in slurry system (not shown here) and the results showed that there was no
catalysis of Ni2+ in solution for photocatalytic reaction. The physical
chemistry properties of the Ni/TiO2 were stable. Little change was observed
even after using for 80 h and the reproducibility of the parallel experiments was tested
to be satisfactory (less than 5%).
Unfortunately, PVA is also a sacrificial electron donor [26].
Thus it reduces the number of photoactive substances for its competition reaction to OH·
or positive hole. Therefore, it is not a stable and good binder in practical applications.
We have ever used polytetrafluoroethylene (PTFE) as a binder but the results were not
satisfactory due to its poor performance of binding. It appears to be necessary to probe
more suitable binders and this is under way in our laboratory.
3 CONCLUSIONS
It has been shown that it was possible to destroy PCA over TiO2 immobilized on
porous nickel by binder in photocatalytic reaction. This is a fairy simple method and
potential used as immobilized catalysts working in batch photoreactor from the point of
application with advantage of eliminating the final filtration of TiO2 fine
particles in suspension. In the absence of oxygen, external anodic potential bias can make
photocatalytic reaction proceed. These may provide a quite desirable approach to degrade
organic substrates. It is worthy to point out that photocatalysis produces more toxic
photoproducts such as nitrobenzene and 4-chloronitroaniline during the reaction, so it
needs to take measures to totally eliminate the photoproducts in its applications.
REFERENCES
[1] Hoffman M R, Martin S T, Wonyong C et al. Chem. Rev., 1995, 19: 69.
[2] Oills D F. Environ. Sci. Technol., 1985, 19: 480.
[3] Matthews R W. J. Phys. Chem., 1987, 91: 3328.
[4] Preis S,
Krichevskaya M, Kharchenko A. Wat. Sci.
Technol., 1997, 35: 265.
[5] Matthews R W,
Mcevoy S R. Solar Energy., 1992, 49:
507.
[6] Vinodgopal K,
Hotchandani S, Kamat P V. J.
Phys. Chem., 1993, 97: 9040.
[7] Hidaka H, Asai Y,
Jincai Z et al. J. Phys. Chem.,
1995, 99: 8244.
[8] Pelegrini R,
Peralta-Zamora P, Andrade A R D
et al. Appl. Catal. B: Environ., 1999, 22: 83.
[9] Butterfield I M,
Christensen P A, Hamnett A et al. J.
Appl. Electrochem., 1997, 27: 385.
[10] Byrne J A, Eggins
B R. J. Electroanal. Chem., 1996, 457:
61
[11] Fernandez, Lassaletta G, Jimenez V M et al. Appl. Catal. B: Environ., 1995, 7: 49.
[12] Liu Hong, Cheng Shao'an, Zhang Jiangqing et al. Chemosphere, 1999, 38: 283.
[13] Brillas E, Bastida R M, Uosa E. J. Electrochem. Soc., 1995, 142: 1733.
[14] Hand R L, Nelson
R F. J. Electrochem. Soc., 1978,
125: 1059.
[15] Pramauro E, Prevot
A B. Analyst, 1995, 120: 237.
[16] Pruden A L, Ollis
D F. J. Catal., 1983, 82: 404.
[17] Daniel J W. Analyst, 1961, 86: 640.
[18] Analysis method for environmental pollutants, Vol.1: inorganic
substance (in Chinese). 1987. Beijing: Kexue Press.
[19] Claire R, Boule P. J. Photochem Photobiol A: Chem., 1991,
60: 235.
[20] Ken-ichi Okamoto, Yasunori Yamamoto, Hiroki Tanaka et al. Bull. Chem.
Soc. Jpn., 1985, 58: 2015.
[21] Prevot A B,
Vincenti M, Bianciotto A et al. Appl.
Catal. B: Environ., 1999, 22: 149.
[22] Abdullah M, Lew G
KC, Matthews R W. J. Phys. Chem., 1990,
94: 6820.
[23] Lew G KC, MeEvoy S
R, Matthews R W. Environ. Sci.
Technol., 1991, 25: 460.
[24] Herrmann J M, Mozzanega M N,
Pichat P. J. Photochem., 1983, 22: 333.
[25] Sánchez L, Peral J, Domènech X.
Electrochim. Acta, 1997, 42: 1877
[26] Brown G T, Darwent J R, Fletcher P D I. J. Am. Chem. Soc., 1985, 107: 6446