http://www.chemistrymag.org/cji/2001/038039ne.htm |
Aug. 1,
2001 Vol.3 No.8 P.39 Copyright |
Polyethylene glycol as support and phase transfer catalyst in aqueous palladium-catalyzed cross-coupling reactions
Xia Min, Wang Yanguang
(Department of Chemistry, Zhejiang University, Hangzhou 310027, China)
Received Feb.21, 2001; Supported by the National Natural Science Foundation of China(29972037)
Abstract Excellent yields and purity were
obtained in the aqueous palladium-catalyzed Suzuki, Sonogashira, Still and Heck reactions
under mild conditions using polyethylene glycol (PEG) as soluble polymeric support and
phase transfer catalyst as well.
Keywords liquid-phase synthesis, polyethylene glycol, cross-coupling
reaction, phase transfer catalyst, palladium catalyst
The construction of libraries of compound
using combinatorial and parallel synthesis has witnessed dramatic developments in recent
years[1]. Insoluble polymers of all kinds have thus been examined as possible
supports, providing significant advantages over many conventional solution phase routes.
However, such an approach requires a great deal of developmental time and efforts to work
up synthetic conditions on solid supports since generally slower kinetics and variations
in behavior. Furthermore, it is always difficult in following reactions and characterizing
products still attached to the polymer by routine analytical methodologies (e.g. 1H,
13C-NMR, I.R., TLC, etc.)
The use of soluble polymers, i.e. polymers which are soluble in one
medium but insoluble in another and purity can therefore be carried out through
precipitation by filtering and washing away unwanted materials after each operation, have
overcome the drawbacks of working on insoluble supports to great extent. While the
research on the preparation of peptides, olignonucleotieds and oligosaccharieds on soluble
supports has been extensive, the synthesis of small molecules, especially the synthesis by
transitional metal catalyzed reactions, has been very few [2,13]. So far,
polyethylene glycol (PEG) appears to be the most promising and versatile soluble polymer
in liquid phase synthesis[3].
It was reported that iodobenzoate and iodophenol could be carried out
cross-coupling reaction in water without organic cosolvent under phosphine-free palladium
catalysts4, thus avoiding phosphine-related side reactions[5].
Aqueous reaction conditions offer a safe, economic and environmentally benign alternative
in organic synthesis, but are often limited by spare solubility of reactants in water. In
some reactions related to "ligandless"
palladium catalyst, phase transfer catalyst (PTC) is necessary to carry out reactions
under mild conditions and increase the yields[6]. Furthermore, PEG has been
investigated as thermally stable, recoverable, inexpensive water-soluble and nontoxic PTC,
presumably operating by the same mechanism as crown ether[7]. Here, we report
the aqueous palladium-catalyzed Suzuki, Sonogashira, Still and Heck reactions using PEG as
soluble polymeric support and phase transfer catalyst as well.
We have observed that no additional PTC was needed when 4-iodobenzoate
bound immobilized PEG 4000 was coupled with sodium tetraphenylborate (1), aryl
boronic acids (2), aryltributylstannanes (4) alkenyl (5) and alkynyl
compounds (3) in water. The reactions required lower temperatures and shorter time
with improvement of yields compared with those analogous reactions in solution phase 8-11,14,
utilizing the intrinsic solubility and phase transfer catalytic properties of the
polymeric support. Only very recently the combined use of PEG as polymeric support and PTC
was reported[12,13].
It was found that aryl iodides gave incomplete conversion in water with
organoboronic acids under palladium acetate by tetrabutylammonium bromide as PTC[14].
In our reaction, however, due to the intrinsic catalytic activity of the polymeric
support, the cross-coupling reaction of PEG 4000 bound 4-iodobenzoate with phenylboronic
acid (2a) or 4-methylphenylboronic acid (2b) was carried out at 60 for 2
hours in excellent yields and purity in the presence of Na2CO3 (Table
1, entry 2,3). Because the reactant of sodium tetraphenylborate (1) was
excessive, the PEG 4000 bond 4-iodobenzoate was converted nearly quantitatively into pure
product (entry 1) in the absence of the additional base (Scheme 1).
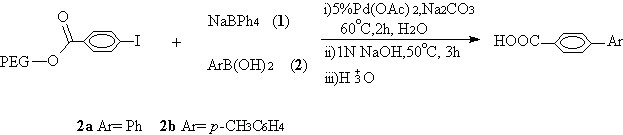
Scheme 1
Bumagin et al reported that aryl halides were carried out the Sonogashira reaction with terminal acetylenes in water under the co-catalyst of Pd(PPh3)2Cl2/CuI in the presence of 10% Bu3N [9]. The catalytic amount of tributylamine was used as indispensable PTC. In our case, the cross-coupling reaction of terminal acetylenes (3) with PEG bound 4-iodobenzoate under Pd (OAc)2/CuI in water could be taken place at room temperature in excellent yields and appropriately quantitative purity without any additional PTC (Scheme 2,Table 1,entry 4,5,6).
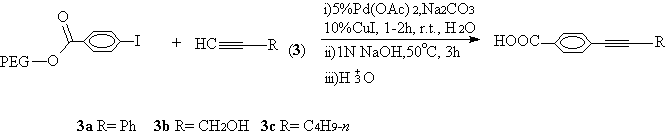
Scheme 2
Normally, the reaction temperature for Still reaction is high. Thanks to the "cuprous effect", this reaction can undergo under mild conditions at the help of catalytic CuI [10]. When phenyltributylstannane (4a) and 4-methylphenyltributystannane (4b) coupled with PEG bound 4-iodobenzoate in water in the presence of Pd(OAc)2/CuI, the reaction could take place at room temperature within 1 hour in high yields and purity (Scheme 3,Table 1, entry 7,8).
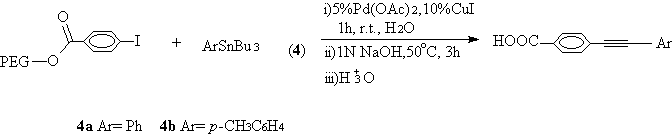
Scheme 3
Water was found to be an effective medium for Heck reaction[8,11]. However, the yields were quite low without equal amount of PTC such as quaternary ammonium salts. Due to the PTC property, PEG bound 4-iodobenzoate could react with styrene (5a), propenol (5b) or acrylic acid (5c) in water without additional PTC in excellent yields and purity (Scheme 4, Table 1, entry 9,10). However, it appeared that electronic-withdrawing carboxyl group had electronic effect on the coupling of acrylic acid (5c) with PEG bound 4-iodobenzoate in relatively low yield and purity (Table 1, entry 11). This result is in accordance with Bumagin's report that long time was needed and low yield was obtained in the case of the reaction of acrylic acid with diaryliodonium salts bearing electronic-withdrawing groups in water under Pd(OAc)2 as catalyst[15].
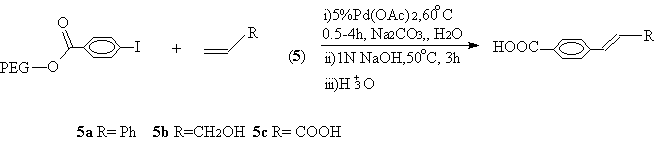
Scheme 4
Although known reducing agent such as tertiaryl phosphine[16] were not present under "ligandless" conditions in this system, the above all reaction mixtures darkened and palladium metal began to deposit soon after the addition of the catalyst and immediately upon heating. This indicated that Pd (II) was readily reduced by some other constitute in the mixture. Pd (0) is not known to be able to exist in solution in the absence of strong donor or acceptor ligands[17]. PEG has been described as PTC presumably operating by the same mechanism as crown ether[7]. It seems to be likely that PEG is able to stablize the Pd (0) species. It was reported that the "ligandless" cross-coupling reactions with PEG as PTC could also proceed under heterogeneous procedure on palladium metal[18]. All these show that PEG acts double roles as soluble support and PTC as well.
Table1 Palladium-catalyzed reactions on PEG bound 4-iodobenzoate in water a, g
Entry |
Substrate |
Time (h) |
Temp. (°C) |
Observed MS (%) |
Crude |
Crude |
|
1c |
1 | 2 |
60 |
198 |
198 |
89 |
99.18 |
2 |
2a |
2 |
60 | 198 | 198 |
92 |
99.43 |
3 |
2b | 2 |
60 | 212 |
212 |
86 |
98.88 |
4b |
3a |
1 |
r.t. |
222 |
222 |
91 |
98.13 |
5b |
3b |
2 |
r.t. | 131 |
176 |
84 |
98.34 |
6b |
3c |
2 |
r.t. |
129 |
202 |
87 |
98.97 |
7b,c |
4a |
1 |
r.t. |
198 |
198 |
90 |
99.17 |
8b,c |
4b |
1 |
r.t. |
212 |
212 |
86 |
98.65 |
9 |
5a |
0.5 |
60 |
105 |
178 |
83 |
94.33 |
10 |
5b |
2 |
60 |
179 |
224 |
83 |
90.39 |
11 |
5c |
4 |
60 |
148 f | ----- |
71 |
75.74 |
The PEG bound products
were subjected to a very efficient cleavage from the support with 1N NaOH aqueous solution
at 50 for 3 hours. The process could be monitored by TLC with the disappearance of the
original point. Through extraction with either after acidification under 5N HCl aqueous
solution to pH 3~4, the crude products were determined with purity more than 90% by GC
analysis (Table 1).
In summary, we have shown a liquid-phase methodology for the
palladium-catalyzed cross-coupling reactions in water on soluble support of PEG 4000. The
reaction not only provide corresponding products in excellent yields and purity, but also
were carried out under mild conditions due to the contribution of PEG bound substrates as
PTC. It is potential for these reactions to be assumed for the combinatorial and parallel
synthesis on the soluble polymeric support.
EXPERIMENTAL
General procedure for the preparation of PEG bound 4-iodobenzoate: at
room temperature, 2g PEG 4000 (with loading capacity of 0.5mmol/g), 4-iodobenzoic acid
(0.496g, 2mmol), dicyclohexylcarbodiime (DCC, 0.412g, 2mmol) and N,N-dimethylaminopyridine
(DMAP, 0.024g, 0.2mmol) were dissolved in anhydrous CH2Cl2 (10mL)
and the resulting mixture was stirred at 37°C overnight. After
filtration, the filtrate was precipitated with ether, the resulting white solid was washed
with ether for several times and dried over vacco to provide the PEG bound 4-iodobenzoate
in quantitatively.
Typical procedure for the preparation of entry 6 is as follows: at room
temperature, 1.5g PEG bound 4-iodobenzoate, 1-hexyne (0.123g,1.5mmol) and sodium carbonate
(0.16g, 1.5mmol) in 8mL water were injected into the mixture of palladium acetate
(0.0182g,0.075mmol) and cuprous iodide (0.029g,0.15mmol) under nitrogen. The resulted
solution was stirred at room temperature for 2 hours. The water was coevaporated with 2mL
toluene at 60°C under reduced pressure. Then toluene (10mL)
added and the mixture centrifuged. The clear supernatant was precipitated with 50mL cold
ether and filtered, washing with ether for three times. The crude PEG bound product was
redissolved in 5mL dichloromethane, the precipitated and washed with cold ether for three
times, drying in vacco for the next sequence.
The saponification of PEG bound product was representative as below: 1.4g
PEG bound 4-hexynl benzoate was dissolved in NaOH aqueous solution (1N, 15mL) and heated
at 50°C for 3 hours (tested by TLC until the original point
disappeared). After cooled to room temperature, the solution was acidificated with HCl
aqueous solution (5N) to pH within 3~4. By extracting with ether (2×5mL),
the combined organic layer was dried over anhydrous sodium sulfate and given the crude
product 4-hexynl benzoic acid as yellow solid (0.131g, 87%). The crude purity of this
compound was determined to be 98.97% by GC analysis. 1H-NMR(500MHz,CDCl3)
0.97 (t,3H),1.49 (h,2H),1.61 (p,2H),2.45 (t,2H), 7.71(d, J=8.4Hz,2H), 8.02 (d, J=8.4Hz,2H)
FT-IR(KBr) 2953, 2670, 2252, 1688, 1606, 1425, 1281, 1120, 908, 732 MS m/z (%) 202
(45), 187(45), 159 (51), 142 (18), 129 (100), 115 (56), 77(14)
REFERENCES
[1] Creswell M W, Bolton G L, Hodges J C et al. Tetrahedron, 1998, 54: 3983.
[2] Blettner C G, Konig W A, Stenzel W et al. Synlett., 1998, 295.
[3] Wentworth P, Janda K D. Chem Commun., 1999, 1917.
[4] Bumagin N A, Bykow V V, Beletskaya I P. Bull Acad Sci. USSR. Div. Chem. Sci.(Engl.
Transl. ), 1990, 38: 2206.
[5] O'Keefe D F, Dannock M C, Marcuceio S M. Tetrahedron Lett.,
1992, 33: 6679.
[6] Jeffery T. J.Chem.Soc.Chem.Commun., 1984, 1287.
[7] Gokel G W, Goli D M, Schultz R A. J. Org. Chem., 1983, 48: 2837.
[8] Jeffery T. Tetrahedron Lett., 1994, 35: 3051
[9] Bumagin N A, Sukhomlinaova L I, Luzikova E V et al. Tetrahedron Lett., 1996, 37: 897.
[10] Liebeskind I S, Fengl R W. J.Org. Chem., 1990, 55: 5359.
[11] Hiroshige M, Hauske J R, Zhou P. Tetrahedron Lett., 1995, 36: 4567.
[12] Sauvagnat B, Lamaty F, Lazaron R et al. Tetrahedron Lett., 1998, 39: 821.
[13] Blettner G C, Konig W A, Stenzel W et al. J. Org. Chem., 1999, 64: 3885.
[14] Badone D, Baroni M, Cardamone R et al. J.Org. Chem., 1997, 62: 7170.
[15] Bumagin N A, Sukhomlinova L I, Banchikov A N et al. Russ. Chem. Bull., 1992, 41:
2130.
[16] Amatore C, Carre E, Jutand A M et al. Organometallics, 1995, 14: 1818.
[17] Maitlis P M, Espinet P, Russele M J H. In comprehensive Organometallic chemistry,
Wilkinsion G, Stone F G A, Abel E W. Eds. Pergamon: Oxford,1982,Vol. 6, 243.
[18] Andersson C M, Hauberg A. J. Org. Chem., 1988, 53: 235.