http://www.chemistrymag.org/cji/2003/053024pc.htm |
Mar. 1, 2003 Vol.5 No.3 P.24 Copyright |
(China Academe of Engineering Physics, Mianyang 621900; #College of Chemistry &Chemical Engineering Chongqing University, Chongqing 400044, China)
Abstract This paper presents four calculation models of CO adsorbed in surface of TiO2 (110) by applying modeling chemistry,Geometry optimization、 energy calculation and Mulliken population analysis have been researched by ab initio method.The calculation results show that the molecular of CO adsorbed through the atomic of C directly connecting with the molecular of TiO2 is the mainly adsorption manner. The possible mechanism of oxidation reaction of CO has also been researched, comparing with free CO, the reaction activation energy reduces greatly due to nanometer TiO2 adsorbing and the reaction is catalysed.
Keywords nanometer titanium dioxide, chemistry adsorption, ab initio, activation energy, photooxidation
纳米TiO2光催化氧化CO的量子化学研究
熊忠华
魏锡文# 郑秀梅#(中国工程物理研究院,绵阳,621900;#重庆大学化学化工学院,重庆,400044)
2002年12月18日收稿。 摘要 本文运用模型化学的方法,提出CO在金红石型TiO2(110)表面垂直吸附的四种计算模型,并应用ab initio对吸附模型进行构型优化、能量计算及Mulliken集居数分析。计算结果表明,CO在金红石型TiO2(110)表面垂直吸附将以C端吸附为主。在此吸附模型基础上研究了CO被氧化成CO2的可能机理,与自由CO相比,由于纳米TiO2的表面吸附作用大大降低了该反应活化能,从而催化该反应的发生。
关键词 纳米TiO2,化学吸附,从头计算,活化能,光催化氧化 TiO2化学性质稳定、难溶无毒,在有机合成、光解水、环境治理[1,2]等领域显示广阔的应用前景,纳米TiO2由于表面效应、体积效应和量子尺寸效应已经被人们广泛应用于空气和水中污染物的降解。CO是一种常见的大气污染物之一,利用纳米TiO2 对其进行光催化氧化已有许多报道[3,4],但有关该氧化反应机理的量子化学研究尚未见报道。本文采用模型化学的方法首先模拟CO在纳米金红石型TiO2(110)表面的垂直吸附,提出了O--C--Ti的吸附模型,然后通过量子化学的从头计算方法研究了CO在纳米TiO2 表面的氧化机理。 1.计算模型和计算方法
金红石型TiO2晶体属四方晶系,4/mmm点群,其晶体结构如图1所示,晶体参数取自文献[5]。
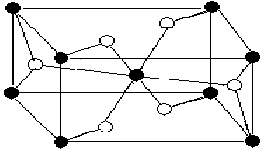 |
晶体参数: ∠OTiO=81.12° ∠TiOTi=98.88° dTi-o(equatorial)=1.947nm dTi-o(apical)=1.982nm |
图1 金红石型TiO2晶体结构 |
|
分别选取簇模型
[TiO5CO]和[Ti5O9CO]模拟CO在TiO2(110)表面的垂直吸附,其中后一种簇模型充分考虑中心Ti原子周围次近邻原子的影响从而更接近真实晶体。因为CO可以分别以C端和O端向TiO2表面吸附,因此选取如图1四种对称型匹配的模型进行吸附研究,运用量子化学的从头计算优化吸附模型的几何构型并计算其能量和电荷分布。采用Gaussian98程序包[6]进行构型优化,原子簇的边界采用Schierbaum提出的“悬挂氢”方案。Ti原子采用Hay和Wadt的带Ar核赝势的(3s2p5d)/(1s1p1d)基组;CO中的C和O原子采用Van Duijneveldt的(9s5p)/(4s3p)基组;表面簇中O原子采用CEP-3G基组;H原子6--31G基组,优化时固定表面簇中的Ti--O键长和∠OTiO、∠TiOTi键角的大小,只对吸附模型中C端吸附时的C--Ti和C--O键长以及O端吸附时的O--Ti和O--C键长,同时对簇模型的能量进行了MP2相关能校正。全部计算均在 Pentium Ⅳ工作站上完成。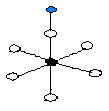 Model
1 C 端吸附 Model
1 C 端吸附 |
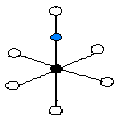 Model 2 O端吸附
Model 2 O端吸附 |
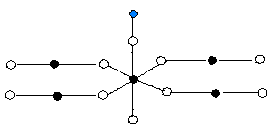 Model 3 C端吸附 |
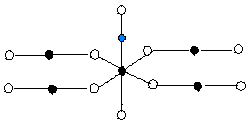 Model 4 O 端吸附 |
图
2 四种吸附模型示意图Fig 2 The structure of four adsorption models 2.计算结果及讨论
2.1 吸附簇模型的从头算优化
表1给出了优化获得的CO垂直吸附于TiO2(110)表面四种吸附模型的一些几何参数和能量。从C原子和O原子之间的键长R(C--O)比较,C端吸附模型Model 1和Model 3中键长明显大于CO分子中键长,而O端吸附模型Model 2和Model 4中键长略大于CO分子中C原子和O原子之间的键长;另外从吸附热考虑,四种吸附模型都呈现不同程度的放热,相比之下C端吸附模型Model 1和Model 3比O端吸附模型Model 2和Model 4放热更多。从以上计算和分析结果可知C 端吸附将比O 端吸附具有更低的能量状态,从而在能量上更为有利,据此我们认为CO分子在TiO2表面的平衡吸附将以C端吸附模型为主。
表2为自然键轨道(NBO)的计算结果,由计算结果可知Model 1和Model 2中C原子和O原子之间的成键轨道(BD(1)和BD(2))中轨道的组成基本相似;但从自然轨道Mulliken集居数分析,Model 2中C--O电子集居数明显大于Model 1中C--O电子集居数;从键能上考虑,Model 2中C--O键能明显大于Model 1中C--O键能。以上计算结果显示,Model 2中C原子与O原子之间的电子云分布较多,说明其键的共价性较强。这与前述构型优化后C--O键的平衡键长是一致的。另外吸附模型Model3和Model4的计算结果亦显示了与Model 1和Model 2较为一致的结论。根据以上的计算及分析结果可知,CO分子垂直吸附于TiO2表面时,在表面Ti原子和周围原子的共同作用下C原子和O原子之间的键长变长,而C端吸附模型键长变化更为显著,键能减小,从而更有利于CO分子被TiO2表面吸附的其它氧化活性物种进一步氧化。
表 1 吸附模型的从头算优化
Table 1 The ab initio optimized of adsorption cluster
Compound |
E(eV) |
R (C--O)/nm |
Adsoption heat(eV) |
CO |
-113.021 |
1.151 |
/ |
[TiO5] |
-1213.031 |
/ |
/ |
[Ti5O9] |
-2990.1 |
/ |
/ |
Model1 |
-1340.966 |
1.289 |
-14.914 |
Model2 |
-1332.475 |
1.204 |
-6.423 |
Model3 |
-3128.383 |
1.432 |
-25.168 |
Model4 |
-3115.707 |
1.343 |
-12.492 |
Table 2 The NBO analysis of adsorption model
Compand |
Orbitals Coefficients |
Occupancy |
Energy(eV) |
||||
| C(s) | C(p) | O(s) | O(p) | ||||
Model 1 |
BD (1) |
0.3501 | 0.6499 |
0.3883 |
0.6117 |
1.9698 |
10.2816 |
| BD(2) | 0.0000 | 1.0000 | 0.0000 | 1.0000 | 1.9627 | 28.5328 |
|
Model 2 |
BD (1) |
0.2303 | 0.7697 |
0.5587 |
0.4413 |
1.9958 |
36.6194 |
BD (2) |
0.0000 | 1.0000 |
0.0000 |
1.0000 |
1.9823 |
29.8085 |
|
Model 3 |
BD (1) |
0.3814 | 0.6186 |
0.2785 |
0.7215 |
1.9979 |
-36.6411 |
BD (2) |
0.0000 | 1.0000 |
0.0000 |
1.0000 |
1.9955 |
-20.3755 |
|
BD (3) |
0.0000 | 1.0000 |
0.0000 |
1.0000 |
1.9951 |
-20.4408 |
|
Model 4 |
BD (1) |
0.0000 | 1.0000 |
0.0000 |
1.0000 |
1.9877 |
-19.4126 |
BD (2) |
0.0000 | 1.0000 |
0.0000 |
1.0000 |
1.9869 |
-19.4453 |
|
BD (3) |
0.2940 | 0.7060 |
0.1643 |
0.8366 |
1.9840 |
-36.4670 |
|
2.2 CO
分子光催化氧化机理纳米TiO2的光催化氧化主要基于当受到能量大于其禁带宽度的紫外光照射时,价带上的电子跃迁到导带上从而导致光生载流子的生成。当光生载流子跃迁到TiO2表面时,与吸附的H2O、OH-等发生反应生成OH·[7]等氧化还原活性物种,进一步参于被吸附物的氧化反应。CO在TiO2表面吸附氧化的可能机理为:OH·首先进攻反应物中(图3所示Reactant)的C原子形成如图3所示的中间产物(Intermediate),然后中间产物失去H·生成如图3所示的产物(Product)和CO2。
化学方程式如(Ⅰ)所示:
Ti-C-O+OH·
为了研究吸附对氧化过程产生的影响,我们同时考察了自由CO分子和OH·的氧化反应,化学方程式如(Ⅱ)所示:
C-O+OH·
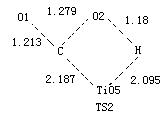
对上述(Ⅰ)和(Ⅱ)两个反应分别用从头计算UB3LYP方法[8-9]优化获得了反应物、过渡态及产物的构型(见表2)。反应(Ⅰ)中第二步基元反应涉及Ti―C解吸、O2―H断裂及Ti―H生成,经过四元环的过渡态(TS2)如上图3所示。其中求过渡态基于负本征值跟踪法[10],经振动频率分析优化所得各反应物和产物均为势能面上的能量极小点,无虚频;各过渡态仅有一个虚频,且经虚振动模式分析加以确认。图4 给出了从头计算方法优化获得的反应(Ⅰ)和(Ⅱ)的反应物、过渡态及产物能级图。两个反应的能量均以各自的反应物能量为零点,图中的ΔE为反应活化能。(表中键长单位均为纳米)
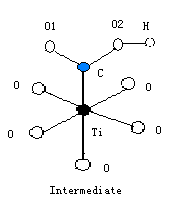
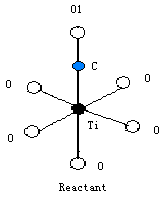
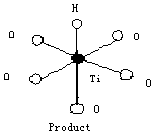 +CO2
+CO2
图
3 CO 氧化反应机理Fig 3 The mechanism of oxidation reaction of CO 表3 反应物、过渡态和产物的从头计算结果
Table 3 The ab initio calculation results of reactant,transition state and productant
| R(Ti--C) | R(C --O1) |
R(C-O2 ) |
A(O1CO2) |
E (eV) |
||
| Reaction(Ⅰ) | Reactant |
1.92 | 1.289 |
/ |
/ |
-1416.694 |
TS1 |
1.98 | 1.290 |
1.375 |
158.4 |
-1416.617 |
|
Intermediate |
2.05 | 1.292 |
1.430 |
119 |
-1416.701 |
|
TS2 |
2.187 | 1.213 |
1.279 |
138.4 |
-1416.697 |
|
Product |
/ | 1.160 |
1.160 |
180 |
-1416.713 |
|
| Reaction(Ⅱ) | Reactant |
/ | 1.151 |
/ |
/ |
-189.099 |
TS1 |
/ | 1.152 |
2.161 |
152.851 |
-188.982 |
|
Intermediate |
/ | 1.155 |
1.329 |
120 |
-189.062 |
|
TS2 |
/ | 1.157 |
1.245 |
147.8 |
-189.051 |
|
| Product | / | 1.160 |
1.160 |
180 |
-189.142 |
|
根据从头算结果,我们可以发现在基元反应(
1)过渡态中C--O1键长有所增长,C--O2键开始形成,并且OH·以一定角度进攻C原子,形成中间产物Ti--COOH,在基元反应(2)中C--O1及C--O2键长均缩小,二者键长趋于平均化,夹角增大。从能级图上我们看到基元反应(1)形成过渡态TS1需克服较大的活化能,分别为2.0944 eV和3.1824 eV(约为202.163 KJ/mol和307.183KJ/mol),可归因为C--O1键长变长以及O原子之间电子云排斥所致。但比较反应(Ⅰ)和(Ⅱ),前者应为表面TiO2的吸附造成C--O1键长相对较长,降低了反应能垒,使反应相对容易进行;基元反应(2)形成过渡态TS2只需克服较小的活化能,分别为0.1088 和0.2992 eV(约为10.502 KJ/mol和28.858KJ/mol)。反应(Ⅰ)和(Ⅱ)相比较,表面TiO2的吸附导致了O--H键长有所增加,降低了反应活化能,使得反应容易发生。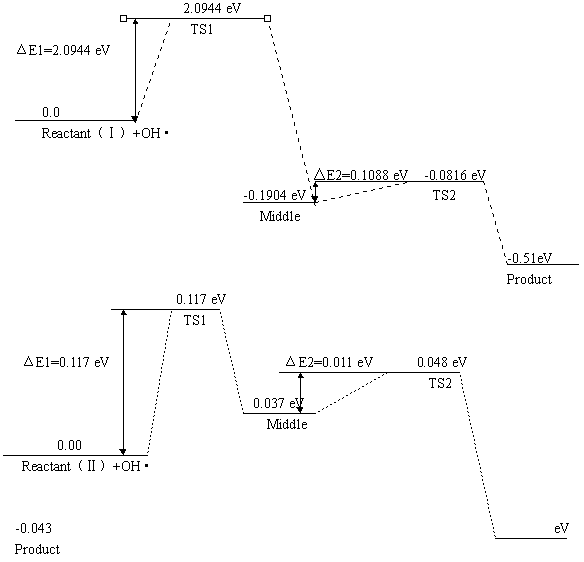
图4 反应(Ⅰ)和(Ⅱ)的反应物、过渡态及产物能级图
Fig. 4 The reactant、transition state and product energy level of reaction (I) and (II)
3 结论
本文通过四种簇模型模拟CO在TiO2(110)表面的垂直吸附,量子计算的结果表明CO在TiO2表面的吸附将以C端吸附为主。CO的吸附氧化机理研究表明,相比自由CO分子的氧化由于TiO2表面的吸附降低了反应能垒,使反应易于进行。
[1] Wu W, Herrman J M, Pichat P. Catal., 1989, 3: 73.
[2] Hoffmann M R, Martin S T, Choi W et al. Chem Rev., 1995, 95: 69.
[3] Fahmi A, Minot C, Silvi B. Phys Rev B., 1993, 47: 18.
[4] Lee W, Shen H S, Dwight K. J Solid State Chem., 1993, 10: 6.
[5] Xu Weixing, Schierbaum K D, Goepel W. J Solid State Chem., 1995, 5: 119.
[6] Frisch M J, Trucks G W, Schlegel H B. Gaussian 98. Pittsburgh: Gaussian Inc1998.
[7] Suzuko Y N, Salvador C M, Kenzi H. J Chem Phys., 1995, 99: 15814.
[8] Becke A D, J Chem Phys., 1993, 98: 5648.
[9] Lee C, Yang W, Parr R G. Phys Rev B., 1988, 37: 785.
[10] Bannerjee A, Adams N, Simons J. J Chem Phys., 1985, 75: 2800.