http://www.chemistrymag.org/cji/2003/057052ne.htm |
Jul. 1, 2003 Vol.5 No.7 P.52 Copyright |
Synthesis of N,N,N',N'-tetra-butyl-3-oxa-pentanediamide and its analogous compounds
Zhang Ping, Chen Jing, Li Chunyu, Tian Guoxin
(Institute of Nuclear Energy Technology, Tsinghua University, P.O.Box 1021, Beijing
102201, China)
Received May 6, 2003; Supported by the National Natural Science Foundation of China
(No.20106009).
Abstract Four diamides have
been synthesized by a four-step reaction in our laboratory. They are
N,N,N'N'-tetra-butyl-3-oxa-pentanediamide (TBOPDA), N,N,N'N'-tetra-(2-methylpropyl)-3-oxa-
pentanediamide (TMPOPDA), N,N,N'N'-tetra-hexyl-3-oxa-pentanediamide (THOPDA), and
N,N,N'N'-tetra-(2-ethylhexyl)- 3-oxa-pentanediamide (TEHOPDA), respectively. The
purification method of the product has been studied. The products were identified with
FT-IR, 13C-NMR, ESI-MS and elemental analysis.
Keywords Diamide, Synthesis, Purification
The safe treatment and disposal of the radioactive waste, specially high level waste(HLW), from nuclear power plant is crucial to the sustainable development of nuclear energy. The partitioning and transmutation of long-life nuclides such as minor actinides from HLW is a method to reduce the long-term risk of HLW. In the studies of HLW partitioning, the first task is to develop novel extractants. During the last 20 years, the solvent extraction of actinides and fission products by monoamides and diamides has been widely studied [1-5]. Compared with organophosphorus extractants, the diamides derivatives have some remarkable advantages. First, they have a high irradiation stability and a strong affinity to metallic ions such as actinides in acidic solutions. Secondly, they are completely incinerable without secondary solid waste because they consist of C, H, O and N elements. The above advantages imply that the diamide derivatives are promising in the treatment of radioactive waste.
Diamides can be generally expressed as (RR'NCO)2R''. In the structure of diamides, the nature of the three groups R, R' and R'' plays an important role on the coordination ablility of the extractant. A kind of diamide derivatives named oxapentanediamide, was widely studied because of its desirable coordination properties of oxygen to metallic ions. The synthesis method was reported by Pretsch[6] and Thiollet[7] as follows:
(C4H9CH3NCO)2CH2 + LiC4H9 = C4H10 + (C4H9CH3NCO)2CHLi
(C4H9CH3NCO)2CHLi + R(OC2H4)nBr =(C4H9CH3NCO)2CH (C2H4O)nR
The main disadvantage of this method is that the required alkyl substituted ether halide is not commercially available and it is not convenient to prepare in laboratory. Sasaki[8] synthesized diamide derivatives with C6H13 group from diglycolic anhydride. However, his synthesis method was time consuming. For example, some step of the synthesis reaction lasts a week. In addition, the purification of diamide products is difficult. In this study, a mild synthesis technology and a simple product purification method were developed. We obtained the extractants with a suitable lipophilicity and satisfactory extraction ability by changing the substituted groups.
2. SYNTHESIS OF THE DIAMIDE
DERIVATIVES
The synthesis of N,N,N'N'-tetra-butyl-3-oxa-pentanediamide (TBOPDA) and its
analogous compounds consists of the following four steps: (1) diethylene glycol is
oxidized to prepare diglycolic acid; (2) diglycolic acid is converted into diglycolic
anhydride by dehydration; (3) diglycolic anhydride reacts with diamine to give
mono-substituted amide; (4) mono-amide is further substituted to prepare diamide. The
reaction formula is expressed as follows:
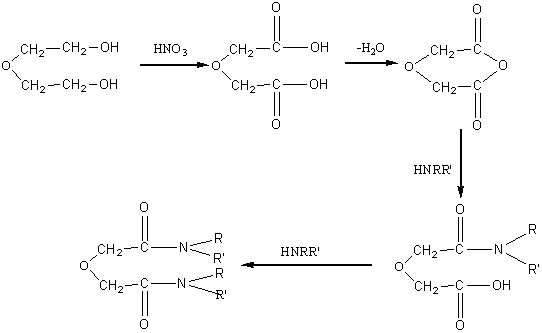
The synthesis is described in detail with TBOPDA as an example.
2.1 Preparation of diglycolic acid
Amounts of 450 mL concentrated nitric acid and 200 mL deionized water were introduced in a
1000 mL reaction vessel with a thermometer, a magnetic stirrer, reflux duct, and recovery
equipment for the acidic gas. Amounts of 4 mL diethylene glycol were added to initiate the
reaction. A great deal of brown gas occurred when the temperature of reaction mixture went
up to 90
The surplus nitric acid was distilled. A small quantity of deionized water was added when brown gas occurred. This process was repeated to remove most nitric acid. Finally, the residue was crystallized and the crystal was then separated, washed with aether, and dried. The product was obtained in a yield of 81%. Its melting point was determined to be 136-139ºC.
2.2 Preparation of diglycolic anhydride
Amounts of 50 g dried diglycolic acid was dissolved in 140mL acetic anhydride. The drops of phosphorus acid were added in as an initiating agent. The temperature rose to the boiling point of acetic anhydride and the reaction mixture was stirred for 1 hour under this condition. Acetic anhydride was evaporated when the temperature rose up to 125ºC. The residue was recrystallized from toluene after most solvent was evaporated out.
The yield is about 90%. The melting point of product diglycolic anhydride is 91-93ºC。
2.3 Synthesis of mono-substituted oxa-pentaneamide
Amounts of 58.5 g diglycolic anhydride and 450 mL 1,4-dioxane were added into a 1000 mL reaction vessel. The mixture was stirred to become transparent. The mixture of 87.5 ml dibutyl amine and 40.6 mL pyridine was added in dropwise under the condition of ice water bath. And then, the reaction mixture was stirred for 3 hours. The 1,4-dioxane is distilled and the crystal appeared when 1:1 hydrochloric acid was dropped into the residual solution. The crystal was separated by filtration and dried and the product was found to be melted at 82-85ºC. The yield is evaluated to be 87%.
2.4 Synthesis of TBOPDA
Amounts of 67 g dried mono-substituted amide (product from step 3) was dissolved in 34 mL thionyl chloride in the reaction vessel under stirring and heating. When the mixture was cooled down to room temperature, 600 mL aether was added under the intensive stirring. The reaction vessel was cooled down to below 0ºC with ice bath. Subsequently, the mixture of 100 mL dibutyl amine, 50 mL pyridine and 40 mL aether was slowly added into the reaction vessel. The reaction lasted for 3 hours after the ice bath was removed. The residue was filtered and the filtrate was alternately washed with 1 mol/L hydrochloric acid and deionized water twice. The organic phase was dried over anhydrous sodium sulfate.
3. PURIFICATION OF
N,N,N'N'-TETRA-BUTYL-3-OXA-PENTANEDIAMIDE AND ITS ANALOGOUS COMPOUNDS
Four diamides with different substituted alkyl (butyl, 2-methylpropyl, hexyl, and
2-ethylhexyl) were synthesized. However, no effective purification method was reported so
far. The common methods, vacuum distillation and over two-time column chromatography[7]
are time consuming or inefficient. A new purification method was developed in this study.
It is an effective purification method for various oxapentanediamides. The purification of
N,N,N'N'-tetra-butyl-3-oxa-
pentanediamide was illustrated as an example.
The crude product was dissolved in petroleum ether. The solution was
mixed with surplus 5 mol/L nitric acid in funnel. The diamide and nitric complex formed
and appeared as a third phase between the organic phase and the aqueous phase. The third
phase was separated, washed with petroleum ether three times, and then the nitric acid in
the complex was neutralized with 2 mol/L NaOH solution. The diamide was dissociated. The
organic phase was washed with 1 mol/L NaOH-H2O-ethanol solution three times to
remove the mono-substituted amide, and then washed with deionized water to remove the
residual ethanol and NaOH solution.
The organic phase was dried over anhydrous sodium sulfate. The purified
product was obtained after solvent evaporation. The purity was found to be > 98% with
gas chromatograph, and the yield is about 40%.
4. IDENTIFICATION OF THE PRODUCTS
The purified products were identified with FT-IR (Nicolet 470), 13C-NMR (Bruker
200P FT), Electrospray Mass Spectrum (ESI-MS, PE SCIEX API 3000), and elemental analysis
(C-Flash EA 1112). Their purities were determined with GC-MS (HP 5937).
The detailed results are given as follows:
TBOPDA:
Purity >98%
IR: 1650 cm-1 (nC=O), 1119 cm-1(
13C-NMR (CDCl3):
d12.7(1C), 18.8-19.0(2C), 28.5-29.8(3C), 44.3-45.5(4C), 167.3(5C), 67.8(6C)
MS: 357(M+H)+, 379(M+Na)+
Elemental analysis: C 67.2%;H 11.5 %. N 7.57 %. Calcd. C 67.4 %, H 11.3%, N 7.86%
N,N,N',N'-tetra-(2-methylpropyl)-3-oxa-
pentanediamide (TMPOPDA):
Purity >98%
IR: 1646 cm-1 (nC=O), 1112 cm-1(nas C-O-C), 1040 cm-1(ns C-O-C),1427 cm-1(n-CH2-C=O), 1387 cm-1,
1367 cm-1(d-CH(CH2)2-)
13C-NMR (CDCl3):
![]()
d20(1C), 26-27(2C), 53-55(3C), 170(4C),
68(5C)
MS: 357(M+H)+, 379(M+Na)+
Elemental analysis: C 67.2 %, H 11.4 %, N 7.97 %. Calcd. C 67.4 %, H 11.3%, N 7.86%
N,N,N',N'-tetra-hexyl-3-oxa-pentanediamide
(THOPDA):
Purity >95%
IR: 1646 cm-1 (nC=O),
1119 cm-1(nas C-O-C), 1040 cm-1(ns C-O-C),1427 cm-1(n-CH2-C=O), 1299 cm-1,
1258 cm-1 (nC-N)
13C-NMR (CDCl3):
![]()
d12.8-12.9(1C), 21.4(2C),
30.4(3C), 25.3-25.5 (4C), 26.4-27.7(5C), 44.6-45.7(6C),
167.3(7C), 67.9(8C).
MS: 469 (M+H)+, 491 (M+Na)+
Elemental analysis: C 70.4 %, H
12.2 %, N 6.18%. Calcd. C 71.7%, H 12.0%, N 5.98%
N,N,N',N'-tetra-(2-ethylhexyl)-3-oxa-pentanediamide
(TEHOPDA):
Purity >95%
IR: 1652 cm-1 (nC=O),
1114 cm-1(nas C-O-C), 1040 cm-1(ns C-O-C), 1427 cm-1(n-CH2-C=O), 1262 cm-1
(nC-N), 1227 cm-1,
940 cm-1(nCH(-CH2-)3)。
13C-NMR:
![]()
d13.8-13.9(1C), 22.7-22.9(2C), 28.2-28.6(3C), 30.1-30.3(4C),
36.4-37.6(5C), 23.4-23.5(6C), 10.3-10.7(7C), 44.6-45.7(8C),
169.3(9C), 68.9(10C)
MS: 581 (M+H)+, 603 (M+Na)+
Elemental analysis: C 73.7 %, H
12.5 %, N 4.98%. Calcd. C 74.4%, H 12.5%, N 4.82%
REFERENCES
[1] Cuillerdier C, Musikas C, Hoel P et al. Sep. Sci. Technol., 1991, 26 (9): 1229.
[2] Chen W J, Zhu L, Ding S D et al. Chem. J. Chinese Universities, 1998, 19: 1724.
[3] Nigond L, Musikas C, Cuillerdier C. Solv. Extr. Ion Exch., 1994, 12 (2): 297.
[4] Stephan H, Gloe K, Beger J, et al. Solv. Extr. Ion Exch., 1991, 9 (3): 435.
[5] Nakamura T, Miyake C. Solv. Extr. Ion Exch., 1995, 13 (2): 253.
[6] Pretsch V E, Ammann D, Osswald H F et al. Helvetica Chimica Acta. 1980, 63: 191.
[7] Thiollet G, Musikas C. Solv. Extr. Ion Exch., 1989, 7 (5): 813.
[8] Sasaki Y, Choppin G. R. Anal. Sci., 1996, 12: 225.