http://www.chemistrymag.org/cji/2004/063017re.htm |
Mar.21,
2004 Vol.6 No.3 P.17 Copyright |
A review of desulfurization of light oil based on selective oxidation
Zhao
Dishun, Sun Fengxia, Zhou Erpeng, Liu Yan
(College of Chemistry and Pharmaceutical Engineering, Hebei University of
Science and Technology, Shijiazhuang 050018, China)
Received Oct. 28, 2003; Supported by
the National Natural Science Foundation of China (20276015)
Abstract Environmental concerns have driven the need to remove
sulfur-containing compounds from light oil. Oxidative desulfurization has been given much
interests recently due to its mild condition and without H2. In this paper, the
mechanism, process, and development of selectively oxidative desulfurization are
reviewed.It also introduced the application of the oxidative desulfurization in different
kinds of light oils. In the end, the authors point out the further research needed to be
worked on in this field.
Keywords Desulfurization light oil selective oxidation
A vast variety of sulfur compounds are
present in light oil. These sulfur compounds can be classified into four main groups:
mercaptans (thiols), sulfides, disulfides and thiophenes. Environmental concerns have
driven the need to remove many impurities from light oil. Sulfur-containing compounds are
of particular interest because of their tendencies to produce precursors to acid rain and
airborne particulate material. Therefore, desulfurization of light oil is extremely
important in the petroleum-processing industry.
Several processes have been proposed in the past to deal with the
problem of removing of these compounds from light oil. The most prevalent and common
industrial process is that of treating the fuel under high temperatures and high pressures
with hydrogen. This process is called hydrodesulfurization (HDS) and has received
extensive attention since its original invention in Germany before the Second World War.
Literature describing this technology is immense, amounting to thousands of patents and
scientific and engineering publications[1].
HDS is a process in which light oil is heated, mixed with hydrogen, and
fed to a reactor packed with a particulate catalyst. Temperatures in the reactor typically
range from 600 to 700 ¨H (315
to 370 oC). At
these temperatures, some or all of the feed may vaporize, depending on the boiling range
of the feed and the pressure in the unit. For heavier feeds it is common for the majority
of the feed to be liquid. Reaction pressures range from as low as 500 psig (pounds per
square inch, gauge) to as high as 2500 psig depending on the difficulty of removing the
sulfur. In the manufacture of light oil such as diesel or jet fuel, pressures higher than
800 psig are common. The feed and hydrogen mixture typically flows downward through the
reactor, passing around and through the particulate catalyst. Upon leaving the reactor,
the mixture of treated fuel and hydrogen flows through a series of mechanical devices to
separate and recycle the hydrogen, remove poisonous hydrogen sulfide generated in the
reaction, and recover the desulfurized product. HDS catalysts slowly lose activity with
useing, and must be removed and replaced every two to three years. As used in large
integrated refineries, HDS is very effective and relatively inexpensive[1].
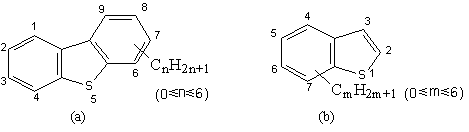
Figure 1. Structure of (a) DBTs and (b)BTs
in actual light oils
However the HDS is
limited in treating benzothiophenes (BTs) and di benzothiophenes (DBTs),especially DBTs
having alkyl substituents on their 4 and / or 6 positions, refer to Figure 1. The
production of light oil, with very low levels of sulfur-containing compounds, therefore
requires inevitably the application of severe operating conditions and the use of
specially active catalysts. An alternative process, able to be operated under moderate
conditions and without urgent requirements for H2 and catalysts is therefore
imminently required[2-4].
Oxidative desulfurization (ODS) has been given much interest as a
further new technology for deep desulfuriztion of light oil recently. This desulfurization
process is composed of two stages: oxidation, followed by liquid extraction. Guth and diaz
(1974) and Guth et al .(1975) disclosed the use of nitrogen dioxides followed by
extraction with methanol to remove both sulfur and nitrogen compounds from petroleum
stocks, Tam and Kittell (1984) described a process for purifying hydrocarbon aqeous oils
containing both heteroatom sulfur and heteroatom nitrogen compound impurities, such as
shale oils, by first reacting the oil with an oxidizing gas containing nitrogen oxides and
then extracting the oxidized oil with solvents in two stages (amines and formic acid), The
oxidation-extraction process used by Patrick et al.(1990) operates at ambient pressure and
low temperature (typically 0-30oC), using nitric acid or nitrogen oxides as oxidants, and
one of several polar solvents for extraction. The oxidized products are composed of a
liquid phase and a byproduct that is a semisolid-like residue with high sulfur content,
hence it reduced sulfur level in the liquid phase[5-9].
Liquid-Liquid extraction is widely used to separate the constituents of
a liquid solution by introducing another immiscible liquid. This process offers improved
product quality and energy savings. In the petroleum industry, solvent extractions have
been used to remove sulfur and /or nitrogen compounds from light oil. The extract oil and
solvent are then separated by distillation. In general, only employing solvent extraction
of petroleum products to remove sulfur creates an oil yield loss, and the sulfur removal
is poor. Such an approach, oxidizing the sulfur compounds of light oil then removing them
by selective extraction, has been achieved success in diesel. However, it has not made
much progress in gasoline due to the high olefins weight content.
It is evident that the greatest advantages of the ODS process are low
reaction temperature and pressure, and that expensive hydrogen is not used in the process.
Another feature of ODS is that the refractory sulfur compounds in ODS are easily converted
by oxidation [10]. Therefore, ODS has great potential to be a complementary
process to traditional HDS for producing deeply desulfurized light oil.
Sulfur-containing compounds are oxidized using a selective oxidant to create compounds that can be preferentially extracted from light oil due to their increased relative polarity. Oxidation is accomplished by contacting an oxidant with light oil under optimum conditions for that light oil and continuing the reaction until oxidized sulfur-containing compounds are confirmed. Oxidation is then stopped before the oxidant attacks other, less reactive, light oil. Or the other parts of the light oil can not be oxidized under such conditions. Light oil containing oxidized sulfur-containing compounds is separated from the depleted oxidant. The oxidant can then be regenerated for re-use. Washing, extracting and chemical post-treatment can remove any unused oxidant that remains in the light oil. The oxidized compounds can be extracted from the light oil by contacting oxidized light oil with a non-miscible solvent. This solvent is selective for the relatively polar oxidized sulfur-containing compounds. The oxidized compounds and solvent are separated from the light oil by gravity separation or centrifugation. The light oil is water washed to recover any traces of dissolved extraction solvent and polished using other methods, such as by absorption using silica gel and aluminum oxide. The extraction solvent is separated from the mixture of solvent and oxidized compounds by a simple distillation for recycling. By following these steps, the highest amount of undesirable compounds is extracted from the fuel while doing the least amount of damage to the end product. In many cases the process improves the fuel quality as well[1,5-9].
Oxidants can convert sulfur-containing compounds to much more polar oxidized species. Such oxidants include peroxy organic acids, catalyzed hydro peroxides, inorganic oxidants such as inorganic peroxy acids, peroxy salts and O3, etc. And such oxidants can donate oxygen atoms to the sulfur in mercaptans (thiols), sulfides, disulfides and thiophenes to form sulfoxides or sulfones, refer to Figure 2. All of these oxidized sulfur-containing compounds are orders of magnitude more soluble in non-miscible solvents than their unoxidized counterparts.
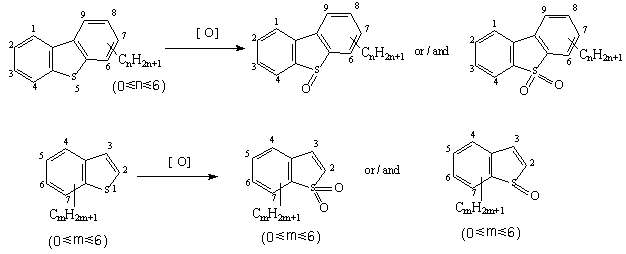
Figure 2. The ideal reaction for DBTs and BTs
The second step of this process is the removal of the oxidized compounds by contacting the distillate with a selective extraction solvent. As reported in the literatures concerning the ODS process[11-15], the liquid-liquid extraction technique using water-soluble polar solvents (DMSO, DMF, and acetonitrile) is usually employed. The former two solvents have a high extractability for sulfones but have a high boiling point at 573K, which is close to the boiling point of the sulfones, and thus they may not be reused for further extraction based on recovery by distillation. In the work of Yasuhiro Shiraishi et al, acetonitrile was used as the extraction solvent, since it has a relatively low boiling point (355K) and is separated easily from the sulfones by distillation. When acetonitrile is contacted with light oil, a large quantity of aromatics is extracted simultaneously with the sulfones. The addition of water however suppresses the extractability of the sulfones [16].
Therefore the solvents should be sufficiently polar to be selective for polar compounds is the process of extraction. Examples of polar solvents include those with high values of the Hildebrand solubility parameter .delta.; liquids with a .delta. higher than about 22 have been successfully used to extract these compounds. Examples of polar liquids, with their Hildebrand values, are shown in the following[1]:
| Acetone | 19.7 |
| Butyl Cellosolve | 20.2 |
| Carbon disulfide | 20.5 |
| Pyridine | 21.7 |
| Cellosolve | 21.9 |
| DMF | 24.7 |
| n-Propanol | 24.9 |
| Ethanol | 26.2 |
| DMSO | 26.4 |
| n-Butyl alcohol | 28.7 |
| Methanol | 29.7 |
| Propylene glycol | 30.7 |
| Ethylene glycol | 34.9 |
| Glycerol | 36.2 |
| Water | 48.0 |
However, as it
will be obvious to those skilled in the arts, mere polarity considerations are
insufficient to define successful extraction solvents. Methanol, for instance, has
sufficient polarity, but its density, 0.79 g/cc, is about the same as that of typical
light oil, making separations very difficult. Other properties to consider include boiling
point, freezing point, and surface tension. Surprisingly, the combination of the
properties exhibited by DMSO make it an excellent solvent for extracting oxidized sulfur
and nitrogen compounds from liquid light oil [1,16,24].
2 DEVELOPMENT
Recent patent disclosures have also
indicated an increased interest in the oxidative approach for sulfur removal. For example,
in U.S. Pat. No. 3,847,800, Guth and Diaz proposed a process for treating diesel fuel that
used oxides of nitrogen as the oxidant. However, nitrogen oxides have several
disadvantages that can be traced to the mechanism by which they oxidize distillates. In
the presence of oxygen, nitrogen oxides initiate a very non-selective form of oxidation
termed auto-oxidation. Several side reactions also take place including the creation of
nitro-aromatic compounds, oxides of alkanes and arylalkanes, and auto-oxidation products.
Oxides of nitrogen are used to synthesize sulfoxides because they tend to inhibit the
formation of sulfones due to the presence of oxonium salts. However, for the purposes of
sulfur removal from light oil, sulfones are the desired product of sulfur oxidation
because of their increased dipole moment, hence, higher solubility in the non-miscible
solvent. Thus, nitrogen oxide based oxidants do not yield the appropriately oxidized
sulfur compounds in distillate light oils without creating many undesirable byproducts.
The Guth and Diaz patent also proposes the use of methanol, ethanol, a combination of the
two, and mixtures of these and water as an extraction solvent for polar molecules.
Although these have been proved to be acceptable extraction solvents for this system, they
do not perform as well as others[17].
U.S. Pat. No. 4,746,420, issued to Darian and Sayed-Hamid also proposes
the use of a nitrogen oxides to oxidize sulfur- and nitrogen-containing compounds followed
by extraction using two solvents--a primary solvent followed by a cosolvent that is
different from the primary[18]. The sulfur and nitrogen results published in
this patent are consistent with those expected from incomplete oxidation of these
compounds followed by extraction.
Tetsuo claims many oxidants as being essentially equal in their ability to oxidize sulfur-
and nitrogen-containing compounds[19]. However many of these oxidants are not
selective and others are ineffective in the later research. Oxidizers that proceed by an
auto oxidation mechanism involving a free radical tend not to be selective for the sulfur-
and nitrogen-containing compounds of interest, producing numerous side reactions and,
hence, various undesirable byproducts.
Tetsuo teaches the use of distillation, solvent extraction, low temperature separation,
adsorbent treatment and separation by washing to separate and oxidized organic sulfur
compound from the liquid oil through the utilization of differences in the boiling point,
melting point and/or solubility between the organic sulfur compound and the oxidized
organic sulfur compound. While most of these work with some success, they do not provide
the level of sulfur removal that his method achieves.
Zannikos et al. describe an oxidation and solvent extraction technique
for the removal of sulfur containing compounds[11]. Peroxyacetic acid was used
in an inefficient manner to oxidize the sulfur compounds in a diesel fuel. Methanol,
dimethyl formamide, and N-methyl pyrrolidone were used as simple one-stage extraction
solvents at different ratios. However, the results of their work show these solvents
removed much of the usable oil along with the oxidized sulfur compounds. In order to get
sulfur levels of approximately 500 PPM with these solvents they report a loss of 30 or
more percent of the overall fuel. Such a loss is completely unacceptable on a commercial
basis. No mention of a process is made within this publication. Instead, the authors
describe laboratory studies of the oxidation and extraction of sulfur compounds using
methods like those taught in the art described above.
Grossman et al. claim a process to remove sulfur from organic compounds and carbonaceous
fuel substrates that contain sulfur chemically bound with carbon [20]. The
process involves a biocatalytic oxidation of the substrates to sulfones and suloxides,
followed by aqueous based desulfurization. In a 1993 European Patent, Funakoshi and Aida
claim a method of recovering organic sulfur compounds from liquid oil using oxidizing
agents, followed by distillation, and solvent extraction or adsorption [9]. The
organic sulfur is recovered as sulfones or sulfoxides. Organic sulfur compounds in fuels
could be effectively recovered by a simple solvent extraction process [21].
Using acetone, dimethylformamide, or other solvents, more than 90% sulfur removal from
various hydrocarbon fuels (ranging from gasoline to straight-run bottoms)could be achieved
through six to eight stages of extractions with a solvent to oil ratio of 1/1 .When an
oxidation step is applied before the extraction , an even higher degree of sulfur removal
is obtained . Earlier work at Alberta Research Council by McFarlane and Hawkins has shown
that organic sulfur in bitumen and synthetic crude oil could be converted to sulfones by
hydrogen peroxide or performic acid although these researchers also have found the
extraction of sulfur compounds from bitumen is ineffective.
Researchers from BP Chemical have reported recently that
dibenzothiophene could be 100% converted to sulfones by using a phosphotungstic
acid/hydrogen peroxide system under mild conditions [3]. Treatment of gas oils
with the phosphotungstic acid/hydrogen peroxide system shows that all the sulfur compounds
present are oxidized. The results also suggest that highly substituted dibenzothiophenes
are the most readily oxidized species containing a thiophenic nucleus. Zannikos et al.
report that a combination of oxidation with solvent extraction is capable of removing up
to 90% of the sulfur compounds in petroleum fractions at acceptable liquid yield [22].
The oxidation process itself leads to substantial sulfur removal without affecting the
boiling point distribution. Dolbear and co-worker report that the more refractory sulfur
compounds could be removed effectively using appropriate oxidants and catalysts at
near-ambient temperature and pressure [23-25]. PetroStar Inc. is one of the
companies that is seriously pursuing this approach [26-28]. Because of their
leading work in this direction, PetroStar has been selected by the US Department of Energy
as one of the three teams to lead the development of ultra-clean fuels by developing new
refining processes that removes sulfur pollutants from crude oil [27].
The desulfurization reactivity of actual light oils, such as
straight-run light gas oil (LGO), commercial light oil (CLO), and light cycle oil (LCO),
of differing sulfur and aromatic concentrations, was studied using oxidant system H2O2
and AcOH, by Yasuhiro Shiraishi et al. The desulfurization efficiency for light oils lies
in the order LGO > CLO > LCO. This is the same as that of the aromatic concentration
in light oils, and demonstrates that high-aromatic-content light oil is difficult to
desulfurize. The sulfones formed by oxidization are not only removed into the aqueous
phase but also form an insoluble precipitate and remain in the light oil. The low
desulfurization efficiency for light oils is caused by the accumulation of sulfones in the
resulting oil[29].
Two main catalysts used for selective desulfurization are organic acid and polyoxometalates. Organic acids include formic acid, acetic acidand so on[1-10, 16,29]. Polyoxometalates have long been studied for oxidation reactions, particularly, the polyoxometalate/hydrogen peroxide system for organic substrate oxidations [30-35]. It has been well documented in the previous oxidation work that the tungsten and molybdenum polyoxoperoxo species in the presence of hydrogen peroxide. The polyoxoperoxo species have been spectroscopically identified and their structures have been determined [36-39]. It was evident that phosphotungstic and phosphomolybdic compounds are catalyst precursors and the actual active species are polyoxoperoxo complexes. Such as, PO4[MO(m-O2)(O2)]43- , PO4[MO(¦Ě-O2)(O2)]23- [33-36].
Limited work has been reported on the detailed mechanistic and kinetic studies for oxidation of organic sulfur compounds in a polyoxometalate/hydrogen peroxide system. One recent example was the oxidation of thioether by polyoxometalate/t-butyl hydrogen peroxide in a non-aqueous system[34]. The Keggin ion, PV2Mo10O405-, was found to be a very active and selective catalyst, which was stable in the redox process. Interestingly, the oxidation reactivities of the representative thioethers in the polyoxometalate/peroxide system do not correlate with their redox potentials, where according to the authors, other factors, such as steric effects, play a significant role.More recently, Otsuki et al. have reported the following trend for sulfur compound oxidation reactivity in a formic acid/H2O2 system [13]: methyl phenylsulfide > thiophenol > diphenyl sulfide > 4,6-dimethyldibenzothiophene > 4-methydibenzothiophene > dibenzothiophene > benzothiophene > thiophenes. This trend confirms that the refractory sulfur compounds in HDS are the most reactive in the oxidation. The reactivities of the compounds seem to correlate well with their electron density except for the dibenzothiophenes with methyl substitutes at 4 and 6 positions[30-36]. Polyoxometalate derivatives catalyze organic substrate oxidations via one of five homogeneous modes [34]; (1) polyoxometalate is then reoxidized by an oxidant, such as hydrogen peroxide; (2) polyoxometalate functions as a cocatalyst, for example, in the Pt/polyoxometalate/O2 system; (3) oxygenation of substrate catalyzed by polyoxometalate derivative; (4) photocatalysis; and (5) the cation of the polyanion activates or oxidizes the sustrate. The polyoxometalate derivatives catalyzed oxidation of organic sulfur compounds discussed in this paper likely takes place with mode 1 or 3.
Mure Te et al. have carried out a comparative study of the dibenzothiophenes oxidations using a series of polyoxometalates as catalyst precursors to better understand the oxidation reactivities of the typical refractory sulfur compounds in diesel fuels [37]. Using toluene solutions of the model compounds of diesel, experiments were carried out to compare the reactivity of the different dibenzothiophenes in oxidation reactions, a key step for oxidative desulfurizations. A series of polyoxometalate/H2O2 systems were evaluated for dibenzothiophene oxidation. The H2O2 solutions of phosphotungstic acid and its salt were very active catalyst systems while their molybdenum counterpart systems were much less active for the model compound oxidation. Oxidation reactivities decreased in the order of dibenzothiophene > 4-methyldibenzothiophene > 4,6-dimethyldibenzothiophene, the same reactivity trend that exists in HDS. However, the oxidation of the dibenzothiophenes was achieved under mild reaction conditions and it was easy to increase reaction temperature or reaction time to achieve high oxidation conversions, even for the least reactive 4,6-dimethyldibenzothiophene. There is a decrease in reactivity of dibenzothiophenes as methyl substitutes increased at the 4 and 6 positions on dibenzothiophene rings. Interestingly, in a formic acid/H2O2 system, the oxidation reactivity of the dibenzothiophenes showed the reverse trend, suggesting that stercoscopic hindrance might play a role when bulky polyoxoperoxo species, which likely form in a hydrogen peroxide solution, act as catalysts[37].
In Yasuhiro Shiraishi et al. paper, the reaction yield for DBTs is increased with an increasing aromatic concentration of the light oil in the order LCOŁľCLOŁľLGO, while that for BTs is decreased in the order LGOŁľCLOŁľLCO. The low desulfurization yield for high-atomatic-content light oil thus results owing to the desulfurization of the BTs being suppressed by the presence of large quantity of aromatics. The remaining percentages for BTs and DBTs tend to decrease with increasing carbon number of substituents. This indicates that the present process desulfurizes the highly substituted BTs and DBTs more effectively. The substitution of hydrophobic alkyl substituents, however, decreases the solubility in the aqueous phase, 26 such that the remaining percentage of sulfones for C0-C3 DBTs and BTs increases with increasing carbon number of the substituents. The cause of the low remaining percentage for C4-C6 sulfones in the case of DBTs and BTs is probably because these compounds precipitate, owing to their low solubility both in light oil and in aqueous solution[29].
Yasuhiro Shiraishi et al. also reported that the desulfurization yields increased with an increasing acetonitrile percentage concentration in the water. The yield lies in the order LGOŁľCLOŁľLCO, which is the same as that for the aromatic concentration in the light oils. The polarity of the light oil increases with increasing aromatic concentration, such that the solubility of the sulfones in the oil also increases with increasing aromatic concentration. As a result, the extractability of the sulfones from high-aromatic-content oil is relatively low. The remaining percentage for the sulfones of both DBTs and BTs, in all the light oils, decrease with increasing carbon number of the substituents. The results show that, in this extraction process, the highly substituted sulfones of DBTs and BTS are removed more easily compared to those sulfones having alkyl substituents of low carbon number. The extractability of sulfones depends on the dipole moment values for the compounds. Highly alkyl-substituted DBT-O2 and BT-O2, of high dipole moment values, is therefore extracted easily from light oil into acetonitrile solution. The proposed process is thus made up of very simple stages and is operated at moderate atmospheric pressure conditions. However, the aromatics in light oils are oxidized to their corresponding carbonyl compounds, which must be removed to improve the combustion property of the light oils[29].
Thus it can be seen that the oxidation and subsequent extraction process shows potential as an energy-efficient desulfurization process for light oil.
4 FURTHER RESEARCH
Two major problems are seen throughout this review. First, the oxidants chosen do not always perform selectively. Many oxidants engage in unwanted side reactions that reduce the quantity and quality of the light oil. The second problem is the selection of a suitable solvent for the extraction of the sulfur compounds. Using the wrong solvent may result in removing desirable compounds from the fuel or extracting less than a desired amount of the sulfur compounds from the fuel. In either case, the results can be costly. So the ODS is still needed to be further researched, especially the catalysts to make the oxidization more selective and detailed mechanism of the above mild oxidization-extraction method REFERENCES
[1] US Patent 6,274,785.
[2] Houalla M, Broderick D,Sapre A V et al. J atal,1980, 61: 523.
[3] Kabe J R,Ishihara A,Jajima. Ind. Eng.Chem. Res, 1992, 31: 1577.
[4] Katzer J R,Sivasubramanian R. Catal. Rev.-Sci. Eng,1979, 20: 155.
[5] US Patent 4493765.
[6] US Patent 4954229.
[7] US Patent 5228978.
[8] US Patent 5458752.
[9] European P Patent 565324-A1.
[10] Collins F M, Lucy A R,SharpC . J Mol.Catal.A: Chem, 1997, 117 : 397.
[11] Zannikos F,Lois E,Stournas S. Fuel Process. Technol,1995, 42: 35.
[12] Aidan T, Yamamoto D. Prepr. Am. Chem. Soc.Div. Pet.Chem, 1994, 39: 623.
[13] Otsuki S, Nonaka T, Takashima N et al. Energy Fuels, 2000, 14: 1232.
[14] Aidan T, Yamamoto D, Sakata K. Trans. Mater, Res.Soc.Jpn,1994, 18A: 39.
[15] Tam P S, Kittrel J R, Eldridfe J W. Ind. Eng.Chem. Res, 1990, 29: 321.
[16]Shiraishi Y,Hirai T,Komasawa.Ind. Eng. Chem. Res. 2000, 39: 2826.
[17] U.S. Pat. No. 3,847,800.
[18] U.S. Pat. No. 4,746,420.
[19] European Patent 93302642.9.
[20] US Patent 5.910.440 .
[21] US patent 5.753.102.
[22] F Zannikos, E Lois, S Stournas. Fuel Proc. Technol, 1995, 42 : 35.
[23] Bonde S E, Gore W, DolbearG E et al. Chem. Soc. Div. Pet. Chem, 2000, 45 (2): 364.
[24] Bonde S E, Gore W, G E Dolbear. Pap. Am.Chem. Soc. Div. Pet. Chem, 1999, 44 (2) : 199.
[25] G.E. Dolbear. E.R. Skov. Prepr. Pap. Am.Chem. Soc. Dov. Pet. Chem, 2000 , 45 (2) : 375.
[26] Whitehurst D D, Isoda T, Mochida I. Adv. Catal,1998, 42 : 345.
[27] US department of Energy. http://www.fe.doe.gov/techline/tl_ultraclean_selection 1.html.
[28] Otsuki S, Nonaka T, Takashima N et al. Energy Fuels ,2000, 14 : 1232.
[29] Yasuhiro, Shiraishi et al. Ind. Eng. Chem. Res,2002, 41: 4375.
[30] Venturello C, Aloisio R D, Bart J C J et al. J. Mol. Catal,1985, 32 : 107.
[31] Ballistreri F P, Tomaselli G A, Toscano R M et al. J: Mol. Catal,1994, 89 : 295.
[32] Salles L, Aubry C, Thouvenot R et al. Inorg Chem,1994, 33 : 871.
[33] Salles L, Aubry C, Robert F et al. J Bregeault. J. Chem, 1993 , 17: 367.
[34] Gall R D, Faraj M, Hill C L. Inorg Chem, 1994, 33 : 5015.
[35] Cavani F. Catal oday,1998, 41: 73.
[36] Girgis M J, Gates B C. Ind. Eng. Chem. Res, 1991, 30 : 2021.
[37] Mure Te, Carig Fairbridge, Zbigniew Ring. Applied Catalysis A: General 2001, 219: 267-280.
ˇˇ ˇˇ