http://www.chemistrymag.org/cji/2005/073024nc.htm |
Mar.20, 2005 Vol.7 No.3 P.24 Copyright |
Liu Hailing1, 2, 3 ,Jiang Huanfeng1* ,WangYugang1,
2, 3 ,Liu Peng1, 2, 3
(
Abstract In this paper,
5-aminolevulinic acid (5-ALA) was simply synthesized from succinyl anhydride via four
steps. In this synthetic technique, we obtained the friend product via the reduction of
nitro group, instead of reduction of acyl cyanide. The total yield is 21.6%. A simple
route for the synthesis of 5-ALA was introduced accompanied with mild reaction conditions,
low toxicity and friendly environment. The structures of all the compounds were confirmed
by 1HNMR, IR and MS.
Keywords 5-aminolevulinic acid(5-ALA);Photodynamic compound; succinyl anhydride
以丁二酸酐为原料合成光动力化合物
5-氨基乙酰丙酸 刘海灵1,2,3,江焕峰1 , 王玉刚1,2,3,刘鹏1,2,3(1华南理工大学化学科学学院 广州 510640;2中国科学院广州化学研究所 广州 510650;3中国科学院研究生院 北京 100039)
2005
年1月31日收稿;国家自然科学基金资助项目(20332030,20172053)摘要 以丁二酸酐为原料经过四步反应简便合成标题化合物5-氨基乙酰丙酸(5-ALA)。本文采用引入硝基再还原得到产物的方法,总产率为21.6%。与文献采用酰氰还原的方法相比,具有试剂廉价易得、毒性低、反应条件温和、环境友好等优点。各步产物均经IR、MS和1H NMR图谱分析确证。
关键词 5-氨基乙酰丙酸;光动力化合物;丁二酸酐
近年来,5-氨基乙酰丙酸(5-ALA)作为一种新型光动力化合物在医学和农业领域具有广泛的应用[1-5]。5-ALA作为一种新型光动力药物,不仅用于局部或全身的皮肤癌的治疗,还可用于膀胱癌、消化道癌、肺癌等的诊断;作为绿色除草剂、杀虫剂、落叶剂、植物生长调节剂等,在农业领域日益受到关注。
有关5-氨基乙酰丙酸合成的文献报道较多[6]。主要有以杂环物质衍生物、b-酮酯类化合物为原料的合成方法。1984年,Pfaltz等人报道了以3-酰氯丙酸甲酯为原料的合成方法[7]。该路线通过引入氰基,再还原水解得到5-ALA。此条路线产率较高,但需用毒性高的CuCN等氰基化试剂,不适合大规模生产。本文从绿色化学的基本原理出发并结合工业化生产的要求,报道了以丁二酸酐为原料,通过合成中间体3-酰氯丙酸甲酯,最终得到标题化合物5-氨基乙酰丙酸的合成方法。该路线反应条件温和、所用的原料试剂价廉易得、毒性低、后处理简便、环境友好、不需特殊仪器设备。合成路线如Scheme 1所示。
1 实验方法
1.1 仪器与试剂
Mercury-Plus 300 核磁共振仪,300MHz,溶剂CDCl3,内标TMS;Analect RFX-65A型红外光谱仪,KBr压片;岛津GCMS-QP5050A气质联用仪,EI源(70ev)。
叔丁醇钾由江阴市新星化工厂提供,工业级;四氢呋喃,经钠回流重蒸后使用;其余试剂均为分析纯。
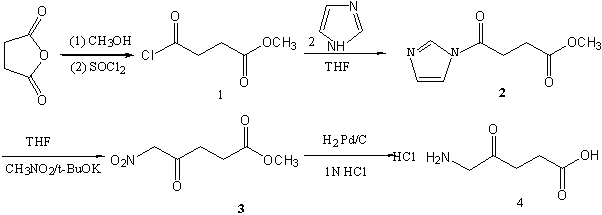
Scheme 1
1.2 3-酰氯丙酸甲酯 1的合成[8]
将20g(0.2mol)丁二酸酐,7.68g(0.24mol)甲醇,加到100mL三口瓶中,搅拌均匀,逐渐升温至90℃。当白色固体完全消失时,停止加热,快速搅拌15分钟,再加热30分钟,得无色透明液体,减压蒸除过量的甲醇。控制温度为30-40℃,逐滴加入47.8g(0.4mol)氯化亚砜,反应3h。减压下将过量的氯化亚砜脱去,在真空度为0.6kPa下,收集79-80℃馏分,得无色液体1 26.4g,产率88.0 %。
IR(cm-1)n:2956,1795,1737,1438,1216,1059。1H NMR(CDCl3,300MHz)d:2.66(t,J=6.6Hz, 2H,CH2),3.21(t, J=6.6Hz, 2H,CH2COCl),3.70(s, 3H,OCH3)。MS:m/z 115(M-Cl),87(100),59,55。
1.2 4-咪唑基-4羰基丁酸甲酯2的合成[9]
将9.0g(0.06mol)3-酰氯丙酸甲酯,50mL四氢呋喃加入150 mL三口瓶中,搅拌均匀,在0℃滴加已溶于50mL四氢呋喃的8.16g(0.12mol)咪唑,滴加完毕后室温搅拌18h后停止反应。过滤、脱溶,冷却后得到浅黄色腊状固体。用四氢呋喃/石油醚纯化得到白色固体8.6g,产率78.8%。
IR(cm-1)n:3143, 2987, 1730,1565,1411,1224,1067。1H NMR(CDCl3,300MHz)d:2.80(t,J=6.6Hz,2H, CH2),3.16(t,J=6.5Hz, 2H,CH2),3.69(s,3H,CH3), 7.08(s,1H,CH),7.44(s, 1H,CH),8.18(s,1H,CH)。MS:m/z 182,151,115(100)。
1.3 5-硝基-4-羰基戊酸甲酯3的合成
在冰水浴中,将1.5g(24mmol)硝基甲烷,20mL DMSO加入100 mL三口瓶中,通氮气,加入溶于40mL四氢呋喃的2.26 g(20mmol)t-BuOK浊液,搅拌10min后,加入溶于20mL四氢呋喃的4-咪唑基-4羰基丁酸甲酯3.50 g(20mmol),室温搅拌36h后停止反应。混合物倒入40 mL蒸馏水中,用乙醚洗涤(20 mL×2),下层水层用1N HCl中和到pH=4-5,用乙酸乙酯或二氯甲烷萃取(50 mL×4),合并有机相,用饱和食盐水洗涤(40 mL×2),无水Na2SO4干燥、过滤、脱溶,得橙色液体1.44 g,产率37.8 %,GC含量92 %。
IR(cm-1)n: 1H NMR(CDCl3,300MHz)d: 2.85-2.71(AA’BB’system,CH2-CH2),3.70(s,3H, CH3),5.37(s, 2H, CH2NO2), MS:m/z 175,144,115(100)。
1.4 5-
氨基乙酰丙酸盐酸盐4的合成将0.35g(2mmol)5-硝基-4-羰基戊酸甲酯, 1N的盐酸10mL,10%的Pd/C0.22g(0.2 mmol)加到30 mL的反应瓶中,常压通氢气,反应24h,停止反应。过滤、旋转蒸发掉溶剂。用乙醇/丙酮纯化后得到棕黄色固体0.27g,产率为81%。
IR(cm-1)n:3126,1725,1403,1178,1049。1H NMR(D2O,300MHz)d:2.59(t,J=6.4Hz,2H,CH2CO2H),2.78(t,J=6.4Hz,2H,CH2)4.02(s,2H, CH2NH3+), MS:m/z 132(100,M-HCl),114,86。(与文献[10]报道一致) 2 结果与讨论
本实验的关键步骤是,硝基甲烷在碱的作用下形成碳负离子,进攻4-咪唑基-4-羰基丁酸甲酯,得到5-硝基-4-羰基戊酸甲酯一步。笔者对该反应的溶剂及碱进行了对照实验,结果见表1。
表1
反应溶剂、碱对反应的影响Table 1 Influence of solvents, bases on the reaction
Entry |
Solvents |
Bases |
Yield % |
1 |
DMSO |
CH3ONa |
32.0 |
2 |
DMSO |
C2H5ONa |
35.4 |
3 |
DMSO |
t-BuOK |
51.5 |
4 |
THF |
CH3ONa |
28.4 |
5 |
THF |
C2H5ONa |
37.8 |
6 |
THF |
t-BuOK |
46.0 |
7 |
CH3OCH3 |
t-BuOK |
35.6 |
从表中可看出:(a)采用二甲亚砜作溶剂,叔丁醇钾作碱,产物的产率最高达到51.5%,粗产物的气相色谱含量为66%。(b)采用四氢呋喃作溶剂,叔丁醇钾作碱,产物的产率为46.0%,粗产物的气相色谱含量最高为92%,可不用纯化直接进行下步反应。二甲基亚砜作溶剂,因其极性高,对碱的溶解性好,有利于反应的进行,但二甲基亚砜的沸点高很难除去,同时它的存在很容易使贵金属催化剂中毒,对下一步采用Pd/C还原硝基有很大影响。因此,本文选用四氢呋喃作溶剂,叔丁醇钾作碱来进行反应。简化了后处理操作,大大减少了溶剂的使用,使成本降低,为5-氨基乙酰丙酸工业化提供了可能。
本文采用Pd/C催化还原硝基,此法与化学方法相比具有环境污染少,可循环利用的优点。Pd/C催化剂价格较贵,使用过程中应该注意Pd/C的回收和活化,使其重复使用降低成本。
本实验以丁二酸酐为原料,通过合成中间体3-酰氯丙酸甲酯,最终成功地合成了目标产物。与Pfaltz等人报道的文献相比,避免使用毒性高的氰基化试剂,采用硝基还原的方法得到5-ALA。四步反应总收率为21.6%。国内关于5-氨基乙酰丙酸的研究刚刚起步,发展新的合成路线及其工艺的工业化是一项十分有价值的工作,值得更多的科学工作者来关注。
[1] Ma J S. Photographic Science and Photochemistry (Ganguang Kexue yu Guanghuaxue), 2002, 20 (2): 131.
[2] Wang J Q, Zhang Z M. (Weishengwuxue Tongbao), 2004, 31 (3): 136.
[3] Rebeiz C A, Constantin A. US 5127938, 1992.
[4]Rebeiz C A, Constantin A. US 5200427, 1993.
[5] Rebeiz C A, Constantin A. US 5321001, 1994.
[6] Liu H LJiang H F. Wang Y G, et al. Modern Chemical Industry (Xiandai Huagong), 2004, 24 (z1): 18.
[7] Pfaltz A, Anwar S. Tetrahedron Letter, 1984, 25 (28): 2977.
[8] Allen F H, Wilson C V. Organic Syntheses, CV3, 169.
[9] Crumbie R L, Nimitz J C, Mosher H S. J. Org. Chem., 1982, 47 (21): 4040.
[10] Nudelman Ayelet, Nudelman Abraham. Synthesis, 1999, (4): 568.