http://www.chemistrymag.org/cji/2005/078056pc.htm |
Aug. 12, 2005 Vol.7 No.8 P.56 Copyright |
Synthesis, characterization of homogeneous Fe-catalyst and its application in selective oxidation of alcohols to aldehydes and ketones
Shen Haoyu1,2*, Zhou Xianbo2, Zhou Saichun2, Mao Honglei2(1 Biological and Chemical Engineering School, Ningbo Institute of Technology, Zhejiang University; 1st Qianhu Nan Road, Ningbo, Zhejiang, 315100; 2Analysis and Testing Center, Ningbo Institute of Technology, Zhejiang University; 1st Qianhu Nan Road, Ningbo, Zhejiang, China 315100)
Abstract The oxidation of alcohols into corresponding aldehydes and ketones is a crucial transformation in organic chemistry, both with academic and industry relevance. Recently, more and more efforts have been put in high valuable catalytic oxidizing process. Among these, protocols based on O2, air or H2O2 are particularly attractive because of cheap and readily available oxidants and with H2O as the only byproduct. In this paper, the synthesis, characterization, and catalytic studies of Fe(III) catalyst, [Fe2O(L)4(H2O)2]2Cl4
·4H2O (1) (L = 6,7,6',7'-Tetramethoxy-[1,1']bi-isoquinolinyl) have been reported for this purpose. The results showed that catalyst (1) can be used for select oxidation of alcohol to corresponding aldehydes or katones. The catalyst (1)-cinnamyl alcohol catalytic system has been monitored by UV-vis before or after adding H2O2 at different time intervals. The result showed the catalytic activity site of the catalyst may be the Fe(III)-O-Fe(III) unit of the catalyst. With reaction time increased, monomer species, Fe(L)32+, was formed as the catalytic oxidation process carring out. When the reaction was carried out more than 24 h, the conversion of alcohol would not be increased. The Fe(III)-O-Fe(III) bridge of the catalyst was fully broken to make the catalyst to be monomer species, Fe(L)32+. Further studies are needed to delineate the intimate mechanistic steps and improve the lifetime of the catalyst.Keywords Selective Oxidation of Alcohols, Homogeneous Fe-catalyst, Synthesis and Characterization, Catalytic Performance, Bi-isoquinolin Ligand. 均相铁(III)催化剂的合成、表征及其在醇的选择性氧化中的催化性能研究
沈昊宇1,2 周先波2
周赛春2 毛红雷2
(1 浙江大学宁波理工学院生物与化工分院 宁波 315100; 2 浙江大学宁波理工学院分析测试中心
宁波 315100)
2005年6月29日收稿;新加坡Agency for Science, Technology and Research (A*STAR)及宁波市科技攻关资助项目
摘要 报道了以m-氧桥(m-O桥)相联的双核铁(III)配合物[Fe2O(L)4(H2O)2]2Cl4·4H2O
(1) (L = 6,7,6',7'-Tetramethoxy-[1,1']biisoquinolinyl; 即6,7,6',7'-四甲氧基-[1,1']双异喹啉)为均相催化剂的设计与合成,研究了它在醇的选择性催化氧化中的催化性质。结果表明,催化剂1对芳香醇和脂肪醇均可以有效地选择性催化氧化为相应的醛或酮。采用UV–vis光谱监测了催化剂---肉桂醇体系在加入H2O2前后不同时间的光谱,结果表明催化剂的活性位可能是Fe(III)–O–Fe(III)。
关键词 醇的选择性催化氧化,均相铁(III)催化剂,合成与表征,双异喹啉配体,催化性能
将醇选择性的氧化成为醛或酮是有机化学中一重要的反应,具有重要的理论研究价值和实用价值[1]。传统方法中,通常采用无机氧化物,如Cr(VI)试剂来实现这一类反应。然而,这类试剂需要计量级投入到反应中;而且通常是有毒有害的,很难从产物中分离;因此是环境不友好也不经济的。更重要的是,大部分这类试剂都难实现多种醇的选择性氧化,特别是那些含有易氧化基团(如不饱和基团或杂原子等)的醇的选择性氧化[2]。近年来,越来越多的科学家致力于高附加值催化氧化过程的研究。其中,采用O2,
空气或H2O2 [3]为氧化剂的研究尤为活跃;这主要是由于这些氧化剂便宜、易得且环境友好。成功的实例包括:Ru-,
Pd-, Cu- 及双金属化合物均相催化剂[4-12]和金属催化剂、以中孔材料,沸石等为载体的异相催化剂等[5-19]。本文报道了以m-氧桥(m-O桥)相联的双核铁(III)配合物[Fe2O(L)4(H2O)2]2Cl4·4H2O
(1) (L = 6,7,6',7'-Tetramethoxy-[1,1']biisoquinolinyl; 即6,7,6',7'-四甲氧基-[1,1']双异喹啉)为均相催化剂的设计与合成,并研究了它们在醇的选择性催化氧化中的性能。
1 实验部分
1.1 主要仪器和试剂
EURO EA elemental analyzer CAP 40/2型元素分析仪; ICP-OES
型等离子发射光谱仪;Bio-Rad EXCAL3000型红外光谱仪; Shimadzu UV-2550
型紫外---可见分光光度计;TA instrument SDT 2960 simultaneous DSC-TGA型热分析仪;Auto
Lab -100电化学分析仪; Agilent HP-6890N气相色谱(HP-5毛细管柱);Agilent
HP-6890N/MS-5793气相色谱----质谱联用仪;所用试剂均为分析纯试剂。配体L的制备方法参照文献[20]。
1.2 催化剂的合成
催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O
(1)的制备方法如图1所示:1:2摩尔比的FeCl3·6H2O
(0.1 mmol, 27.3 mg)和配体 L (0.2 mmol, 75.2 mg)分别溶于10 mL甲醇中,混合后,室温搅拌下滴加Et3N至pH
= 8~9。滤去反应后的不溶物后,得褐色溶液,室温下静置约一周,得到褐色微晶,过滤,用少量水和乙醇洗涤,真空干燥,产率87%
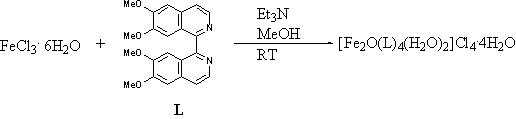
图1 催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O
(1)的制备反应
Fig. 1 Preparation of the
catalyst [Fe2O(L)4(H2O)2]2Cl4· 4H2O (1)
1.3 催化剂的催化活性评价实验
以(1)为催化剂,H2O2为氧化剂,考察了肉桂醇等8种醇的选择性氧化。各种底物的实验过程类似。实验条件及过程以选择性催化氧化肉桂醇为例说明如下:5
g肉桂醇溶于10 mL甲苯中,升温至80oC,搅拌回流下缓慢加入15
mg(0.3 wt%)的催化剂(1),1 mL H2O2。每隔15分钟取样,产物以GC-MS定性,GC定量分析。醇的转化率和产物选择性的计算公式分别为式(1)和(2):
转化率(%)= [(A0-At)/A0]×100% (1)
其中,At:时间为t时的反应物的GC峰面积, A0:时间为t0时的初始反应物的GC峰面积
选择性(%)= [npi/ å npi]× 100% (2)
其中,npi:时间为t时的目标产物的量, å
npi:时间为t时的所有产物的量
2.1催化剂组成及物理性质
催化剂(1)的元素分析实验值(理论计算值)分别为(%):H: 4.88(4.92),C: 56.18 (56.12),N: 5.92 (5.95),O: 19.56 (19.54),Fe: 5.88 (5.93)。以DMF为溶剂测定了配合物的电导,数据表明配合物1:4型电解质。采用AgNO3滴定配合物水溶液体系中的Cl-离子,实验值(理论计算值)分别为(%):Cl: 7.56 (7.53)。配合物热重差热分析表明~100oC 有3.9 %重量失重, 相当于4个水分子, ~120oC 有1.9%重量失重, 相当于2个水分子。结果表明,配合物中有2个水分子处于配合物的内界,以配位键与Fe相联,为配合物的配位水。其余4个水分子处于配合物的外界,为配合物的结晶水;而4个Cl-离子均处于配合物的外界。
2.2 催化剂的红外光谱
催化剂(1)在红外光谱上存在特征吸收峰:~1475, 1415和848 cm-1的强吸收峰可归属于配体的C = C和C = N 的振动吸收。
2.3催化剂的电化学性质
为了研究铁均相催化剂的氧化还原性质,测定了催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O的循环伏安曲线(见图2)。实验采用三电极系统,工作电极为铂丝电极(面积为1 cm2),辅助电极为大面积铂片电极,参比电极为Ag/AgCl。扫描速度为100 mV/s;支持电解质(四丁基六氟磷酸胺,TBAP)的浓度为0.1 mol/L。实验前,通氩气除去电解质溶液中的氧,实验在氩气保护下,在室温下(25 oC)进行。得到了两组氧化还原峰。第一对峰(I)对应于[Fe2O(L)4(H2O)2]4+不可逆还原为 Fe(L)32+的峰;第二对峰(II)对应于Fe(L)32+/ Fe(L)33+的氧化还原峰。
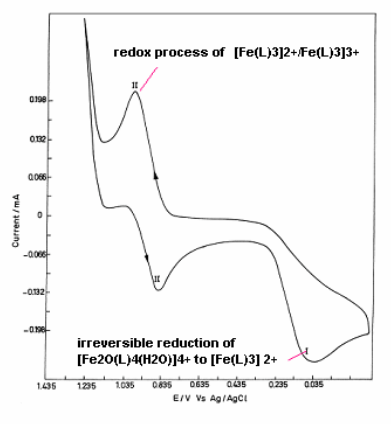
图2 催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O 的循环伏安曲线
Fig.2 The cyclic voltammogram diagram of catalyst [Fe2O(L)4(H2O)2]2Cl4·4H2O
由上述表征推测出催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O可能的结构为由m-氧桥(m-O)桥相联的双核铁(III)配合物,其结构示意图如图3所示。这一结构被下文中2.4.2催化剂的催化性质UV-vis监测中的研究结果是一致的。
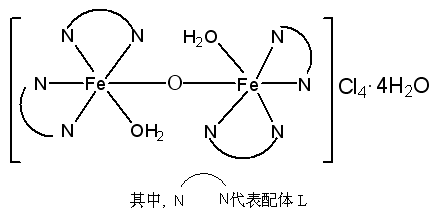
图3 催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O
可能的结构示意图
Fig.3 Presumed structure of catalyst [Fe2O(L)4(H2O)2]2Cl4·4H2O
2.4.1催化剂(1)对8种醇的选择性催化氧化研究
采用GC-MS定性,GC定量考察了以H2O2为氧化剂选择性催化氧化肉桂醇等8种底物的催化反应,结果列于表1。结果表明,催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O对芳香醇和脂肪醇均可以有效地选择性催化氧化为相应地醛或酮。图4中给出了底物2-巯基乙醇以H2O2为氧化剂,时间t = 0和t = 30 min时的GCMS总离子流图和质谱图。在催化剂(1)催化作用下,2-巯基乙醇可以被选择性地氧化成为单一产物2-巯基乙醛。其他各催化剂均可以催化底物选择性地氧化成唯一的目标产物。
表1 [Fe2O(L)4(H2O)2]2Cl4·4H2O对醇的选择性催化氧化研究
(80oC,甲苯,30 min)
编号 entry |
底物 |
产物 |
转化率 |
1 |
|
|
87.5 |
2 |
|
|
88.2 |
3 |
|
|
90.2 |
4 |
|
|
82.7 |
5 |
|
|
83.8 |
6 |
|
|
82.2 |
7 |
|
|
90.2 |
8 |
|
|
91.2 |
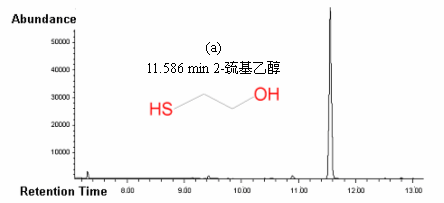
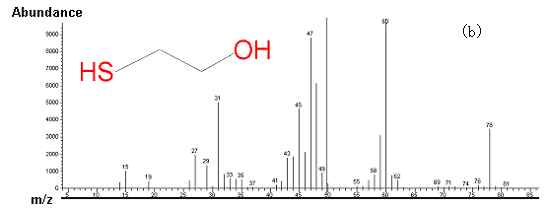
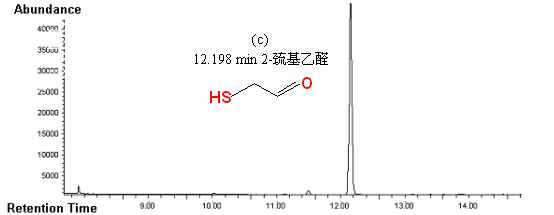
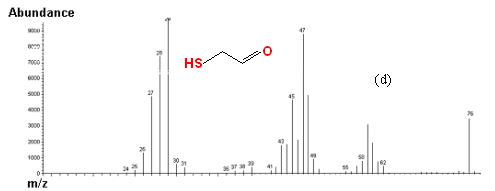
图4 底物2-巯基乙醇以H2O2为氧化剂,催化反应体系的GCMS总离子流图和质谱图
(a)时间t = 0时体系的GCMS总离子流图;(b)时间t = 0时保留时间为11.586
min的组分的质谱图(2-巯基乙醇);(c)时间t = 30 min 时体系的GCMS总离子流图;(d)时间t
= 30 min 时保留时间为12.198 min的组分的质谱图(2-巯基乙醛)
Fig.4 The total ion chromatography (TIC) and
the mass spectrum (MS) of 2-mercaptoethanol and its catalytic oxidation products.
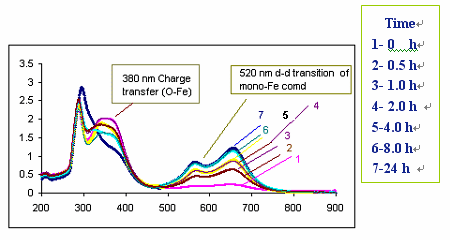
图5 催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O -肉桂醇-H2O2体系的UV-vis光谱
Fig. 5 UV–vis studies of catalyst [Fe2O(L)4(H2O)2]2Cl4·4H2O-cinnamyl alcohol-H2O2 system
2.4.2 催化剂(1)催化性质UV–vis监测
为了观察催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O
的催化性质,运用UV–vis监测了催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O
---肉桂醇体系在加入H2O2前后不同时间的光谱(见图5)。未加H2O2时,在~380
nm 的特征峰可归属为催化剂[Fe2O(L)4(H2O)2]2Cl4·4H2O中氧原子到铁原子轨道(px(O)® dxz(Fe))的电荷转移光谱;这一特征峰是在单核铁化合物的光谱中所没有的。加入H2O2后,~380
nm 的特征峰强度逐渐减弱,而在~520 nm出现一个新的宽峰,而且其强度随时间增长而逐渐增强;表明在催化醇的氧化过程中逐渐形成单核化合物Fe(L)32+。这是由于在H2O2的活化作用下Fe(III)–O–Fe(III)桥被打断而形成单核化合物所致。当反应发生24
h后,醇的转化率不再增加,说明当Fe(III)–O–Fe(III)桥被完全打断而形成单核化合物后则催化剂失效,这意味着催化剂的活性中心为Fe(III)–O–Fe(III)。
3. 结论
以
[1] Larock R C. Comprehensive Organic Transformations. New York: VCH Publishers, 1989.
[2] Sato K, Aoki M, Noyori, R. Science, 1998, 281: 1646.
[3] Ligtenbarg A G, Hage J R, Fering B L. Coord. Chem. Rev., 2003, 237: 89.
[4] Besson M, Gallezot P. Catalysis Today, 2000, 57: 127.
[5] Sloboda-Rozner D, Alsters P L, Neumann R J. Am. Chem. Soc., 2003, 125: 5280.
[6] Khenkin A M, Shimon L J W, Neumann R. Inorg. Chem., 2003, 42: 3331.
[7] Mahata N, Tanielyan A K, Augustine R L et al. Chemical Industries, 2003,89: 551.
[8] Poyatos M, Mata J A, Falomir E et al. Organometallics, 2003, 22: 1110.
[9] Blackburn L, Kanno H, Taylor R J K. Tetrahedron Letters, 2003, 44: 115.
[10] Xu L, Trudell M L. Tetrahedron Letters, 2003, 44: 2553.
[11] Surendra K, Krishnaveni N S, Reddy M A et al. J. Org. Chem., 2003, 68: 2058.
[12] Suzuki T, Morita K, Tsuchida M et al. J. Org. Chem., 2003, 68: 1601.
[13] Zhan B Z, White M A, Sham T K et al. J. Am. Chem. Soc. 2003, 125: 2195.
[14] Shaabani A, Teimouri F, Lee D G. Synth. Commun. 2003, 33: 1057.
[15] Bhar S, Chaudhuri S K. Tetrahedron 2003, 59: 3493.
[16] Griffin K G, Johnston P, Bennett S et al. Chemical Industries, 2003, 89: 169.
[17] Tanyeli C, Gumus A. Tetrahedron Letters, 2003, 44(8): 1639.
[18] Biella S, Rossi M. Chem. Commun. 2003, 3: 378.
[19] Duliere E, Devillers M, Marchand-Brynaert J. Organometallics, 2003, 22: 804.
[20] (a) Busato S, Craig D C, Judeh Z M A et al. Tetrahedron 2003, 59: 461. (b) Siegfried M A, Hilpert H, Rey M et al. Helv. Chim. Acta 1980, 63: 938.