http://www.chemistrymag.org/cji/2006/085035rc.htm |
May 1, 2006 Vol.8 No.5 P.35 Copyright |
Atom Transfer Radical Polymerization and Its Applications
Jiang Zhifei, Zhang Liwu
(College of Chemistry and Chemical
Engineering, Chongqing University, Chongqing 400044)
Abstract In this
paper, the developments of atom transfer radical polymerization (ATRP), included
developments of initiators, catalyzer, and monomer as well as the condition of
polymerization are reviewed. The uses of ATRP to synthesize such as block polymer, star
polymer, graft polymer, hyperbranched polymer, gradient polymer are summarized also.
Keyword Atom transfer radical polymerization(ATRP);block polymer; star polymer; hyperbranched polymer; gradient
polymer
原子转移自由基聚合及其应用
蒋志飞 张立武
(重庆大学化学化工学院 重庆市沙坪坝 400044)
摘要 本文主要综述了原子转移自由聚合反应机理及其引发剂、催化体系、适用单体、聚合条件的发展,并对近几年来利用原子转移自由基聚合在制备嵌段、星型、接技、超支化、梯度等多种拓扑结构的共聚物方面所取得的进展情况作了分析总结。
关键词 原子转移自由基聚合(ATRP); 嵌段共聚物; 星型聚合物;
超支化聚合物; 梯度共聚物
分子设计是高分子制备的一个十分关键的研究内容,设计一种能满足一定需要的高分子材料是高分子化学研究的一项主要目标。活性聚合等高分子合成新方法的出现,为合成结构和分子量可控的聚合物提供了传统聚合方法所没有的手段。但是,活性聚合的反应条件苛刻,工艺过程相对复杂,且现有的活性聚合技术的单体覆盖面较窄,使得分子结构的可设计性较低,从而大大限制了活性聚合技术在高分子材料领域中的应用。而传统的自由基聚合具有单体广泛、合成工艺多样、操作简单、工业化成本低等优点。但是自由基聚合存在与活性聚合相矛盾的基元反应或副反应,使聚合反应过程难以控制。因此,实现自由基的活性可控聚合,是当今高分子合成科学要解决的主要课题之一。原子转移自由基聚合(Atom Transfer Radical Polymerization,ATRP)正是这样一种高分子合成的新技术和新方法[1]。
1 ATRP的反应机理
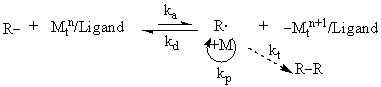
图1 ATRP反应机理
ATRP集活性聚合与自由基聚合特点于一身,因此一经发现得到了广泛的关注。利用ATRP的特点合成出新的聚合物、提高聚合物的性能或改进原来的聚合工艺,降低生产成本是其研究目的也是促进自身发展的动力 [1]。
2 ATRP反应体系的研究进展
引发剂是ATRP研究的一项中心内容,有效的引发剂至少包含一个可进行自由基转移的原子或基团。最初的研究者认为,选择分子结构中含有共轭效应或诱导效应的物质是选择ATRP引发剂的原则,如卤代苯基化合物、羰基基化合物、腈基化合物及多卤代化合物等[3]。随后Percec等发现,含有削弱S-Cl键的取代芳基磺酰氯也可引发苯乙烯和丙烯酸酯类单体聚合[4]。 然而随着研究的深入,董宇平等发现即使分子结构中没有共轭或诱导基团的卤代烷在 Fe·4H2O/PPh3的催化作用下,也可引发甲基丙烯酸丁酯的可控聚合,从而拓宽了ATRP的引发剂选择范围[5]。反向原子转移自由基聚合(RATRP)的成功,引发剂的选择范围更是被大大拓宽。传统的自由基引发剂如过氧化物、过硫化物、偶氮化合物、过氧碳酸盐、过硼酸盐、高氯酸盐、过酸等也被用做RATRP的引发剂 [6]。
2.2 催化体系的发展
催化体系是ATRP成功与否的最重要因素,包括可与引发剂发生可逆性氧化还原反应的过渡金属化合物及可与过渡金属化合物配位的配位体两部分。理想的ATRP催化剂应当有较高的原子转移选择性,不存在副反应,对不同单体应有比较容易调节的活性速率常数。
催化体系的研究第一代ATRP 技术是以卤化亚铜为催化剂,随后报道为Ti、Fe、Co、Ni、Mn、Ru、Nb、Mo、Rh、Pd等过渡金属成功地应用于ATRP催化体系[7]。配体也是ATRP 催化体系的一个重要组成部分,具有增加催化剂的溶解性能和稳定过渡金属的作用。第一代ATRP 技术中配体为联二吡啶,随后又有Patten等人用烷基取代的联二吡啶作为其代替物。现在Haddleton等人开始使用廉价易得的多胺、亚胺等逐渐代替吡啶而作为配体。这些催化剂用于ATRP 反应的相继成功为人们开发高效环保的催化剂树立了信心[7]。
Matyjaszewski等发现使用混合金属体系可增加对ATRP的可控性,混合金属包括铜、铁、锰、铬、镍、银、钯、金、铼、铱、铟、钌、钐、锌、铑等的混合物。在这混合金属体系中,第一过渡金属应有一种更高的氧化态,第二种过渡金属包含一种更低的氧化态,还应当有一种零价的过渡金属。这种混合金属物体系的好处就是可以对分子量分布、聚合速率、引发速率等进行多种控制。此外,通常情况下加入零价过渡金属加入到以相应的过渡金属化合物做催化剂的聚合反应中可以极大地提高聚合速率[7]。
Sawanoto等人报道了以钌(Ⅱ)化合物为催化剂,有机铝化合物为助催化剂,使卤原子顺利转移,这是一种与卤化亚铜不同的催化体系[8]。
2.3 可适应单体的种类
与其他活性聚合相比, ATRP具有更宽的选择范围, 一般分子结构中含有共轭效应或诱导效应的单体都可进行ATRP。主要为烯键式不饱和的单体,包括苯乙烯及其衍生物、(甲基)丙烯酸酯类、含氟乙烯型单体、乙烯基醋酸、异丁烯、丙烯腈、丙烯酰胺、乙烯基吡啶、N-环已基马来酰亚胺、对羧基保护过的丙烯酸等。Patten等研究发现,因不同单体在ATRP聚合中有各自的增长速率常数、自由基浓度、终止速率常数,所以不同单体的原子转移自由基聚合,各自需一套合理配置的引发体系[9]。
2.4 反应条件
对ATRP 反应条件的优化主要是对反应温度和反应介质的优化。低温ATRP一直是人们研究的方向和目标,目前人们主要通过选择合适的催化体系实现温度的优化。Sawamoto等采用RuH2 (PPh3)4 及单茂铁系列催化剂成功地实现了低温ATRP,该催化体系在低温条件下,反应速度不仅没有降低,反而有明显加快[10]。另外,还可选用一些极性单体(如甲基丙烯酸甲酯)在室温下的聚合,反应仍具有“活性”特征[1]。反应介质的选择也是影响反应的一个很重要因素,选择溶剂时必须考虑自由基向溶剂的链转移常数大小、溶剂和催化剂之间的相互作用等方面。最初人们主要使用有机溶剂作为ATRP的反应介质,也有用超临界的二氧化碳用作反应介质的报道[11]。水相ATRP的成功同样引起人们的关注,以水为介质既经济又无污染,必将成为人们研究的热点[1]。近来,具有表面引发体系的ATRP报道,引起了学术界的关注,它以硅胶或玻璃为固定界面,在其表面产生一定结构功能的、具有优良的电化学及机械性能的膜或刷,为分子识别、传感装置、分子设计所需的功能化膜提供了一条新的思路。离子溶液为介质的ATRP体系,具有催化剂脱除容易和抑制副反应发生等优点,更是当前具有挑战性的课题[12]。
3 ATRP的应用
嵌段聚合物、接枝聚合物、星形聚合物、超支化聚合物及多种其它类型聚合物。
3.1 ATRP合成嵌段共聚物
目前利用ATRP法制备嵌段共聚物又可分为全ATRP法和半ATRP法。根据ATRP的反应机理,制得的聚合物一端为卤素原子X的RX,以此它可以作大分子引发剂,重新引发第二单体的ATRP反应,得到不含均聚物、分子量及组成可控的纯嵌段共聚物,这种方法称为全ATRP法(Strictly ATRP)。Matyjaszewski等合成了丙烯酸丁酯(n-BA)-丙烯酸三甲基硅氧乙酯(TMSHEA)两嵌段共聚物,TMSHEA嵌段水解后转化为聚丙烯酸羟乙酯(HEA),最终得到双亲性嵌段共聚物[13]。傅志峰等发现,当用ATRP法合成苯乙烯(St)-甲基丙烯酸甲酯(MMA)嵌段共聚物时,聚合顺序对嵌段效率有很大影响,当先进行MMA 的聚合,然后再进行St 的聚合时,得到了完全的嵌段共聚物;反之,如果改变单体的聚合顺序,则嵌段效率很低,有相当一部分的PSt没有引发MMA 的聚合[14]。
对于某些不能进行ATRP,甚至不能进行自由基聚合的单体,可在带有特定官能团的聚合物末端引入可以引发ATRP聚合的活性卤原子作为ATRP 的大分子引发剂,来制备嵌段共聚物,此法称为半ATRP法。Nakagawa等先将端乙烯基聚二甲基硅氧烷与对二甲基硅氢亚乙基苄基氯反应,得到两端为苄基氯的聚二甲基硅氧烷,用它作为大分子引发剂,CuCl/bpy 作为催化剂,分别与St 和n-BA 进行ATRP,得到了以聚二甲基硅氧烷为中间嵌段的ABA 型嵌段共聚物,这种聚合物集无机/有机性能于一身,具有独特的性质[`15]。Yu等ATRP与活性阴离子聚合相结合,先利用ATRP用2,2-二氯-2?羟基-乙酸乙酯(DCAG)做引发剂,与苯乙烯(PS)聚合,得到含有活性羟基的(Cl-PS) 2-CHCOOCH2CH2OH,经过脱氢、活性阴离子聚合、加成得到(PS)2-PEO-DCA,以其作为引发剂,再利用ATRP在130℃下与甲基丙烯酸甲酯聚合,从而得到H 型的 (PS)2-PEO-(PMMA)2嵌段共聚物[16]。这样可以集各种聚合机理的特点于一体,弥补单一机理之不足,使不同聚合性质的单体能相互结合形成嵌段共聚物。
3.2 ATRP合成接技或梳形共聚物
目前利用ATRP 制备的接技共聚物,主要是“从主干接技” (grafting from) 或“直接接技”(grafting through) 两种方法合成接技共聚物。
“从主干接技”主要是使主链侧基功能化,使其带有多个引发单元,这些引发单元能够引发单体聚合形成接技链。这方面应用最广泛的是通过大分子卤化反应[17]和大分子氯甲基化反应制得接技共聚物[18]。最早的例子是将乙烯基单体接技到聚氯乙烯上形成主链为聚氯乙烯的接技共聚物,这种共聚物玻璃化温度下降,聚氯乙烯的柔性增加[19]。Gao等将间同立构的聚苯乙烯引入溴代乙酰基,以其作为大分子引发剂,利用ATRP分别与甲基丙烯酸甲酯和苯乙烯聚合得到相应的接枝共聚物 [20]。
“直接接技”是指大分子的均聚或共聚形成接技的方法,Matyjaszewski利用“直接接技”的方法,以氯代乙烯基酯引发苯乙烯的进行ATRP,所得大分子再和N-乙烯基吡咯烷酮进行共聚,生成的接技聚合物是一种主干为亲水性的乙烯基吡咯烷酮和酯双键的共聚链,接技链为疏水性的聚苯乙烯的双亲性水凝胶 [21]。刘兵等通过St 与4-对氯甲基苯乙烯(CMS) 进行氮氧稳定自由基共聚合反应, 合成了二元共聚物(PS-co-CMS) , 并以此共聚物引发丙烯酸丁酯进行原子转移自由基聚合, 成功合成了结构明晰的以聚苯乙烯为主链, 聚丙烯酸丁酯为支链的接枝共聚物[22]。
3.3 ATRP合成星型共聚物
一般说来, 星形聚合物制备方法可分为多官能度引发剂法和偶联法两种。原子转移自由基聚合的出现, 使星形聚合物的合成更为简便。利用ATRP,以小分子的多官能团做引发剂合成星形聚合物在ATRP出现后的不久就被人们发现。Matyjaszewski等第一个以六溴甲基苯为引发剂制得六臂星形聚苯乙烯,所得聚合物分子量与理论值相近,分子量分布为1.23[23]。用无机杂环小分子作多官能团引发剂的也有报道,Matyjaszewski 等还报道了分别利用环四硅氧烷和环三叠氮膦为引发剂制备四臂和六臂聚合物,并发现将所得聚合物用空间排阻色谱法(SEC)测得的分子量比理论值低,而用粘度测试法测得的分子量与理论值基本一致[24]。
ATRP的偶联法制备星形聚合物是利用ATRP首先制备得到聚苯乙烯、聚丙烯酸叔丁酯等线性聚合物,然后这种聚合物与二乙烯基苯、1,4-丁二醇甲基丙烯酸甲已二酯等关联剂形成关联中心从而形成星形聚合物[3]。选择卤代物、溶剂、核素、偶联反应速率是其中决定星形聚合物形成的重要因素[25]。Wu等以线形聚苯乙烯(PS)作大分子引发剂,二乙烯基苯(DVB)作偶联剂,制备星形聚苯乙烯(PS)n ,核内残留未反应的乙烯基基团进行氢溴化处理,以此作为大分子引发剂引发丙烯酸乙酯(EA)聚合,从而得到(PS)n-(PEA)m的杂臂星型共聚物,其中PEA的臂数大于PS的臂数[26]。
3.4 ATRP合成超支化共聚物
采用ATRP引发体系引发同时带卤原子和双键的AB*星型双官能团单体(又称之为能发生自缩合作用的乙烯基单体SCVP),可以得到超支化聚合物(也称树枝状聚合物),聚合物的支化度可通过改变AB*型单体的量和聚合反应时间来控制。生成的聚合物端基含有卤原子, 加入其它单体,反应可以继续进行合成出树枝状或超支化嵌段共聚物。卤化物的端基能还能被其它功能基例如叠氮基、氨基、羟基、环氧基等取代,在一定条件下转化为具有某些特定功能高分子[3]。
第一个利用ATRP合成超支化聚合物的例子是对乙烯基苯甲基氯(VBC)单体。Weimer和Gaynor等分别用低浓度(5%)和高浓度(>20%)的催化剂对其进行了研究,结果发现,使用高浓度的催化剂得到的聚合物支化度更高[27]。Cheng等利用ATRP-SCVP将p-氯甲基苯乙烯(CMS)或p-溴甲基苯乙烯(BMS)与2,3,4,5,6-五氟苯乙烯共聚,得到含氟的超支化共聚物,这种反应条件的优点是能够对共聚物组成、分子量、支化度、性质进行控制,因此被广泛用于将含氟单体制备成超支化聚合物的研究中[28]。
3.5 ATRP合成梯度共聚物
采用ATRP合成梯度共聚物方法有两种, 一是用竞聚率差别较大的两种单体一次加料法, 另一种是分阶段加料法。Min等研究了在本体和微乳液体系中同时发生反向ATRP和正向ATRP引发,从而得到丙烯酸丁酯和甲基丙烯酸丁酯的梯度共聚物,共聚物组成从甲基丙烯酸丁酯链节占主导地位逐步变化为丙烯酸丁酯链节占主导地位[29]。Sawamoto等用RuCl2(PPh3)3/Al(OiPr)3为催化剂, 1-PEBr为引发剂, 先加入St/MMA 的组成比为3/1的单体混合物, 在反应过程中通过两次补加MMA , 制备了St/MMA 组成为分别为3/1、1/1、1/4的ABC 型三嵌段共聚物,其相对分子量分布约为1.5,当用钌为过渡金属元素催化剂时聚合物分子量将更高[30]。
4 结束语
[1] Matyjaszewski K et al. United States Patent: 6538091, 2003.
[2] Wang J S, Matyjaszewski K. Macromolecules. 1995, 28 (23): 7901-7910.
[3] Matyjaszewski K, Xia J H. Chem. Rev. 2001, 101(9):2921-2990.
[4] Percec V, Barboiu B. Macromolecules, 1995, 28(23): 7970.
[5] Wang W J, Deng Y P. Chemistry (Gaofenzi Xueshu Lunwen Baogaohui Lunwenji), Part A, 1999: 93.
[6] Matyjaszewski K et al. United States Patent: 6759491, 2004.
[7] Matyjaszewski K et al. United States Patent: 6790919, 2004.
[8] Sawamoto M, Kato M, Kamigaito M et al. Polymer Priprints(ACS Div Polym Chem), 1995, 36 (1): 539-540.
[9] Adam L, David H M. Prog. React. Kinet. Mech. 2004, 29(3):187-242.
[10] Kotani Y, Kamigaito M, Sawamoto M. Macromolecules, 2000, 33(10): 3543-3549.
[11] Xia J H, Johnson T, Scott G et al. Macromolecules, 1999, 32 (15): 4802-4805.
[12] Tadesuz B, Przemyslaw K. Journal of Polymer Science, Part A: Polymer Chemistry. 2002, 40(16): 2799- 2809.
[13] Muhlebach A, Gaynor S G, Matyjaszewski K. Macromolecules, 1998,31:6046-6052.
[14] Fu Z F, Shi Y, Jiao S K et al. Chemistry (Huaxue Xuebao), 1999,6: 692-696.
[15] Nakagawa Y, Matyjaszewski K. Polymer Priprints(ACS Div Polym Chem), 1996, 37(2): 270-271.
[16] Yu Xifei, Shi Tongfei, Zhang Guo et al. Polymer, 2006, 47(5): 1538-1546.
[17] Wang X S, Luo N, Ying S K. Polymer, 1999, 40(16): 4515-4520.
[18] Zhang Y M, Luo N, Ying S K. Chemistry (Gaodeng Xuexiao Xuebao), 1999, 20: 492.
[19] Matyjaszewski K, Beers K L, Coca S et al,. Macromolecules, 1999, 40(2): 95-96.
[20] Gao Yong, Li Songqing, Li Huaming et al. European Polymer Journal, 2005, 41(10): 2329-2334.
[21] Matyjaszewski K, Beers K L, Kern A et al. Journal of Polymer Science, Part A: Polymer Chemistry. 1998, 36(5): 823-830.
[22] Liu B, Hu C P. Chemistry(Gaofenzi Xuebao), 2002, 1: 47-52.
[23] Wang J S, Greszta D, Matyjaszewski K. Polymeric Materials Science and Engineering. 1995, 73: 416.
[24] Matyjaszewski K, Miller P J, Pyun J et al. Macromolecules, 1999, 32(20): 6526-6535.
[25] Zhang X, Xia J, Matyjaszewski, K. Macromolecules, 2000, 33 (7): 2340-2345.
[26] Wu Y, Shi Y, Fu Z F. Polymer, 2005, 46(26): 12722-12728.
[27] Gaynor S G, Edelman S Z, Matyjaszewski K. Polymer Priprints(ACS Div Polym Chem), 1996, 37(2): 413-414.
[28] Cheng Chong, Wooley K L, Khoshdel E. Journal of Polymer Science, Part A: Polymer Chemistry, 2005, 43(20): 4754-4770.
[29] Min K E, Mei L I, Matyjaszewski K. Journal of Polymer Science, Part A: Polymer Chemistry, 2005, 43(16): 3616-3622.
[30] Kotani Y, Kamiqaito M, Sawamoto M. Macromolecules. 1998, 31(17): 5582-5587.