http://www.chemistrymag.org/cji/2006/088052pc.htm |
Aug. 1, 2006 Vol.8 No.8 P.52 Copyright |
Zhao Aiqin, Li Zhanchen#, Dong Zhimin, Dou Lanfeng, Han Xue#
(Min Ke Environmental Detection of limited
company, Baodong 071051; #College of Chemistry and Environmental Science, Hebei
University, Baoding, 071002, China)
Abstract 2- methyl - naphtho- thiazole of
(1, 2-d); (2, 3-d); (2, 1-d), in these three isomeric bodies, because the base position of
naphthalin is different, the outset raw material is different. This study indicate,
inducting the substituting group in the corresponding position, its result is increasing
the synthesis difficulty.This article simultaneously discussed these three kinds of
isomeric bodies' physical and chemical performance.
Keywords 2- methyl - naphtho- thiazole isomer synthesis
2-甲基-萘并噻唑的三种同分异构体的合成 及其理化性能的比较
赵爱琴
,李占臣#,董志民,窦兰凤,韩雪#(河北省保定市民科环境检测有限公司, 071051;#河北大学化学与环境科学学院,保定市 071002)
2006
年4月23日收稿摘要
2-甲基-萘并(1,2-d);(2,3-d);(2,1-d)噻唑,三个同分异构体,由于萘并基的位置不同,导致了起始原料的不同,在相应的位置导入取代基,其结果加大了合成的难度,本文对此进行了研究,同时讨论了三种同分异构体的理化性能。关键词 2-甲基-萘并噻唑 同分异构体 合成 1 前言
2-甲基-萘并(1,2-d)噻唑是感光材料工业中合成光谱增感染料的重要原材料之一,除了2-甲基-萘并(1,2-d)噻唑之外,还有其它两种同分异构体,它们是2-甲基-萘并(2,3-d)噻唑和2-甲基-萘并(2,1-d)噻唑[1]。为了扩大研究这个系列的同分异构体的实际应用效果,我们在2-甲基-萘并(1,2-d)噻唑合成研究的基础上,开始了后两种同分异构体的合成研究。
表1 三个同分异构体的化学名称及其结构式
| 序号 | 化学名称 | 结构式 | 分子量 | 分子式 |
| 1 | 2-甲基-萘并(1,2-d)噻唑 | 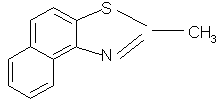 |
199 | C10H8NSCH3 |
| 2 | 2-甲基-萘并(2,3-d)噻唑 | 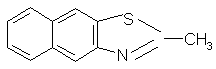 |
199 | C10H8NSCH3 |
| 3 | 2-甲基-萘并(2,1-d)噻唑 | 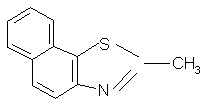 |
199 | C10H8NSCH3 |
本文重点介绍2-甲基-萘并(2,3-d)噻唑和2-甲基-萘并(2,1-d)噻唑两个产品的合成路线的研究以及三个同分异构体的理化性能的比较。
2 合成路线流程图
(1)2-甲基-萘并(2,3-d)噻唑
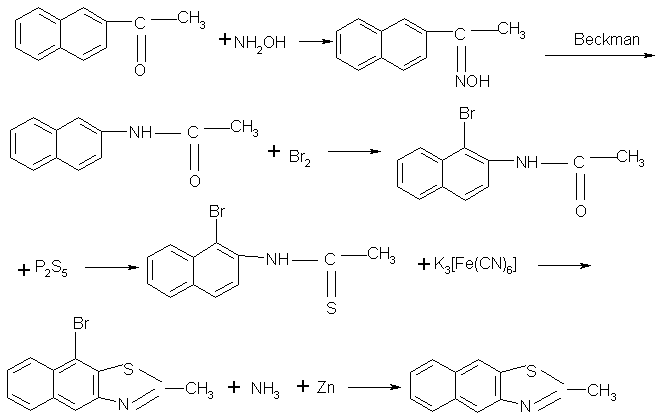
(2)2-甲基-萘并(2,1-d)噻唑
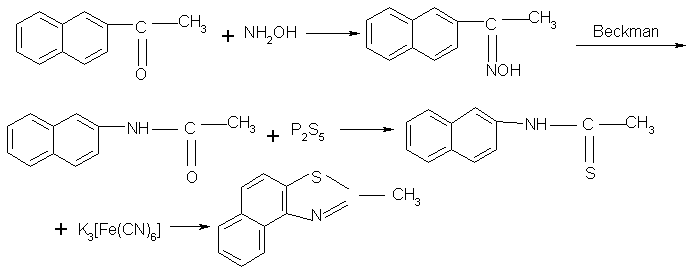
3 合成路线的讨论
3.1起始原料的选择
(1) 2-甲基-萘并(1,2-d)噻唑的起始原料是甲萘胺,经酰化、硫代和氧化闭环得到产品。如果按照产品2-甲基-萘并(1,2-d)噻唑的合成路线,后两种产品的起始原料是乙萘胺,由于乙萘胺是一种强烈的致癌物质,已在国内外禁止生产和使用,所以本着绿色染料化学的原则[2],对合成2-甲基-萘并(2,3-d)噻唑、2-甲基-萘并(2,1-d)噻唑两个产品必须采取其它的合成路线。经查阅有关文献[3],决定采用β-萘乙酮作为起始原料。β-萘乙酮与羟胺生成肟,再采用贝克曼反应,转化成β-乙酰胺基萘。
(2) 对于产品2-甲基-萘并(2,1-d)噻唑来说,β-乙酰胺基萘,按2-甲基-萘并(1,2-d)噻唑产品的合成路线,经硫代氧化得到。
(3) 对于产品2-甲基-萘并(2,3-d)噻唑来说,为了使硫代物氧化闭环反应在(2,3-d)位,在合成路线上萘环的1位导入溴取代基,经硫代、氧化闭环反应,从而使萘并基位于(2,3-d),最终经脱溴反应,得到产品2-甲基-萘并(2,3-d)噻唑。
3.2具体合成方法的介绍
3.2.1 β-乙酰胺基萘的合成
(1)肟化反应:75克β-萘乙酮溶解于甲醇中,在50-60℃加入32.5克盐酸羟胺的水溶液,搅拌15分钟后,加入65克醋酸钠的水溶液,加热回馏30分钟。加入少量的水,冷却至0-5℃,过滤,水洗,干燥,得到肟化物79.9克,收率98%。
(2)贝克曼反应:82.5克肟化物加入到8倍量的多聚磷酸中,搅拌均匀,用蒸汽缓慢加热,待温度升到90℃,停止加热。溶液渐渐稀化,内温自升到90-136℃,冷却,迅速降温到80℃,搅拌下放入到200ml水中,冷却到20℃,保持1小时,离心过滤,水洗得到粗品100克。将粗品溶解于200ml甲醇中,搅拌下滴加入300ml水,结晶析出冷却,过滤,水洗,干燥得产品76.5克,收率93%。
(3)合成中要注意的事项:在贝克曼反应中,a、通蒸汽升温的速度要慢,这样可以使固体在粘稠状的多聚磷酸中搅拌均匀,升温速度太快,容易使反应过于激烈,最终温度难以控制,部分产品炭化,降低产品的收率和纯度。b、搅拌速度要快,(因多聚磷酸在常温下为粘稠物,故用手工搅拌)使物料混合均匀,随着温度的升高,物料变稀,要加快搅拌,使料液均匀受热,反应平稳。
(4)问题与讨论:由于β-乙酰胺基萘是产品2-甲基-萘并(2,3-d)噻唑;2-甲基-萘并(2,1-d)噻唑的起始原料,它的纯度和收率直接影响到下一步的反应。因此,a、在肟化反应中,每一步的反应温度要控制好。b、在贝克曼反应中,升温速度要慢,搅拌速度要快;在后处理时,加入300ml水,前40ml水要慢滴加,物料变为糊状物,再滴入100ml水,然后一次性加入剩余的水。这样得到的产品是均匀的粉末。否则,产品为不均匀的硬块,不易粉碎,不易水洗,不易干燥。
3.2.2 2-甲基-萘并(2,1-d)噻唑的合成
对于2-甲基-萘并(2,1-d)噻唑产品,从β-乙酰胺基萘开始,经过硫代,氧化反应可以得到产品。
(1)硫代反应:干燥的β-乙酰胺基萘50克,加上30克五硫化二磷及40ml吡啶放到反应器中,在室温下开始反应,待内温稳定,再升温到100-110℃,反应1小时。冷却到室温,用室温的8%NaOH溶液溶解产品,过滤除去不溶物,滤液在5-10℃用稀醋酸中和到ph=6-7,过滤水洗,得产品40克,收率73%。
(2)氧化反应:100克硫代物溶于8%NaOH溶液中,搅拌溶解,过滤除去不溶物,待用。在反应器中,330克铁氰化钾溶解于680ml水中,内温不高于40℃。在内温38-40℃时,滴加入硫代物的碱溶液,均匀滴加,时间是1-1.5小时。滴加完毕,冷却过滤,水洗,甩干。晾干后减压蒸馏,产品用9:1的甲醇水溶液重结晶,真空干燥得产品60克,纯度99%以上,收率60%。
(3)问题与讨论:该产品熔点较低,75℃。在甲醇中溶解度较大,不适合用甲醇重结晶法,只适合采用减压蒸馏后,用甲醇:水=9:1的甲醇水溶液重结晶,才能得到较好的晶形和收率。
3.2.3 2-甲基-萘并(2,3-d)噻唑的合成
(1)溴代反应 40克β-乙酰胺基萘在50-60℃溶解于冰乙酸中,在同温下滴加溶有34.6克溴的乙酸溶液,析出结晶后,补加部分乙酸,搅拌2小时后,到入冰水中,过滤产品,水洗甩干。将湿品溶于520ml甲醇中,溶解后,冷却过滤,干燥得产品49克,收率86%。
(2)硫代反应 49克溴代物、24.5克五硫化二磷和11.2ml吡啶放入反应器中,在120-130℃油浴中小心地搅拌成糖状物,在此温度下保温15分钟。冷却到50℃,用7%NaOH溶液溶解,有不溶物,加入活性炭过滤,滤液冷却至0-5℃,用5%的乙酸中和,ph=6-7。得到硫代物,过滤水洗,甩干,干燥,得到32.4克产品,收率60%。
(3)氧化闭环反应 将16.3克硫代物溶解在8%NaOH溶液中,过滤待用。在反应器中,将77克铁氰化钾溶解于定量水中,内温40℃以下,在38-40℃下,把硫代物的碱溶液均匀滴加入反应器中,滴加完毕,同温下搅拌2小时。用75ml氯仿抽取,干燥,过滤,蒸溶剂,并用150ml石油醚温浸泡,加活性炭过滤,重复三次。滤液浓缩,冷却,析出结晶,过滤干燥,得到产品6.48克,收率40%。
(4)还原脱溴 28克氧化物,锌粉60克和乙醇搅拌回馏1小时,加氨水,通氨气,蒸乙醇。加水,用氯仿抽取,干燥,过滤。用石油醚温浸泡,加活性炭脱色,过滤,重复三次。滤液浓缩,冷却,析出产品,过滤得产品11.63克,收率58%,纯度98.5%。
(5)问题与讨论
a.溴代反应,采用乙酸介质进行溴代反应,产品的收率达到资料水平[1]。
b.由于在酰胺基的邻位导入溴代基,对随后的硫代反应,氧化闭环反应的具体操作上带来很大的麻烦,其结果造成了各步和最终产品的收率下降。由以下几点可以说明:
① 硫代物采用甲醇热洗,过滤干燥,得到较为纯净的硫代物。
② 根据我们的试验,利用产品在氯仿中溶解度较大,将中间体4-溴-2-甲基-萘并(2,3-d)噻唑从反应液中提取出来,蒸干氯仿,再用石油醚抽取。因为氯仿抽取出的产品含杂质较多,需用石油醚再次抽取,净化产品。该中间体在石油醚中的溶解度较小,所以用大量的石油醚多次抽取结晶,再浓缩蒸干石油醚,得到棕色固体,直接用于下一步脱溴反应。
③脱溴反应,在通氨气的条件下,用锌粉脱溴还原,虽然延长了通氨气的时间,收率仍然很低,有待进一步提高。
④由于产品的熔点高,145℃,无法按前两种产品进行减压蒸馏和甲醇重结晶。我们采用丙酮盐酸盐法提纯产品,得到纯度99%以上的产品。这种方法用于提纯杂环类产品的效果明显。
4
三种同分异构体理化性能的比较
4.1 熔点
序号 |
化学名称 |
熔点(℃) |
1 |
2-甲基萘并(1,2-d)噻唑 |
97-97.5 |
2 |
2-甲基-萘并(2,3-d)噻唑 |
145-145.5 |
3 |
2-甲基-萘并(2,1-d)噻唑 |
75-75.5 |
由于产品的熔点不同,结果对产品的后处理有所不同。
2-甲基-萘并(1,2-d)噻唑:采用减压蒸馏,再用甲醇重结晶,得到较好的晶形和收率。
2-甲基-萘并(2,3-d)噻唑:熔点高,减压蒸馏的温度也高,产品易分解,采用丙酮提纯法得到很纯的产品。
2-甲基-萘并(2,1-d)噻唑:减压蒸馏得到半成品,如果用甲醇重结晶,因为该产品在甲醇中溶解度较大,产品不易析出,故采用甲醇与水的混合溶剂提纯。
4.2 纯度
采用上述提纯方法,三种产品的纯度在99%以上,完全达到产品指标。
4.3 产品的紫外吸收光谱
光谱图:
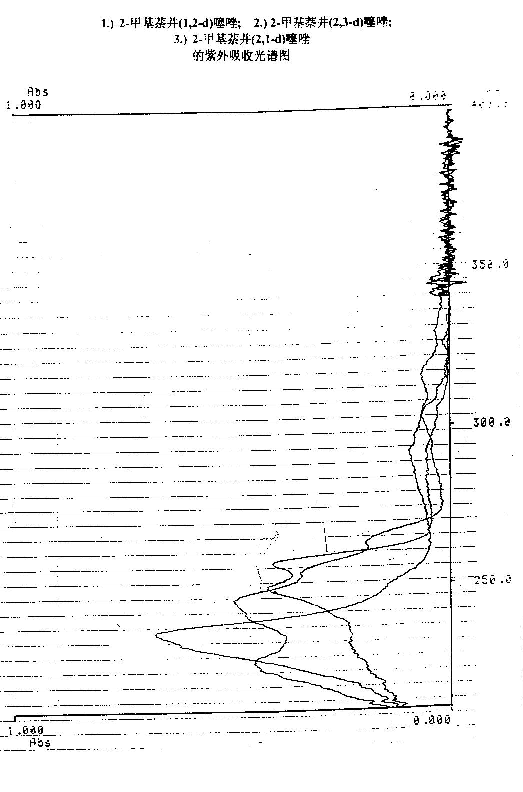
在相同的浓度下,2-甲基-萘并(1,2-d)噻唑:紫外吸收最大值
2-甲基-萘并(2,3-d)噻唑:紫外吸收形成双峰,其吸收强度较低。吸收峰1 lmax=225nm OD=.0450,吸收峰2 lmax=245nm OD=0.497,吸收范围 220-275nm。
2-甲基-萘并(2,1-d)噻唑:紫外吸收形成双峰,吸收强度较低。吸收峰1 lmax=250nm OD=0.420,吸收峰2 lmax=257nm OD=0.408,吸收范围 240-260nm。
由图中可以看出,三个产品在相同的条件下,2-甲基-萘并(1,2-d)噻唑,在235nm处,吸收强度最大,OD=0.677,吸收范围较窄。2-甲基-萘并(2,3-d)噻唑产品在235nm的两侧各有一个吸收峰,它的波长吸收范围较宽,但它的吸收强度较小,OD=0.4左右。2-甲基-萘并(2,1-d)噻唑在235nm的右侧有两个吸收峰,吸收强度OD=0.4。 5 结论
1.三个同分异构体:
(1)由于萘并基位置不同,造成了合成路线的不同,增加了合成难度。
(2)由于其理化性能(熔点,溶解度等)的不同,使得产品的后处理方法有所不同,即使产品的纯度指标达到要求,但其收率也有所不同。
(3)由于产品的紫外吸收光谱有所不同,从而可以清楚地看出,2-甲基-萘并(1,2-d)噻唑属于单一吸收峰,峰形较陡峭,适用于彩色胶片;至于其它两种同分异构体,属于双峰吸收,峰形较平坦,它们可能适用于黑白胶片。
2.整个实验过程符合绿色染料化学的要求,合成工艺中不含禁用物质,因此可以应用于实际生产。 REFERENCES
[1] 新井厚明《高机能ァォトヶミカルス》ミ棩ē啷窏(1986)
[2] 陈荣圻,绿色染化料的开发和应用(一),印染,2006 (6): 45
[3] 唐培堃.中间体化学工艺学.化学工业出版社出版,1984: 37
[4] 闫起强,李承恩,张军. .感光科学与光化学,2000,18(2):170
[5] Morita Kosuke, Ebara Kazuya, Shibasaki Y. et al,New positive-type photosensitive polyimide having sulfo groups,Polymer(Juhewu), 2003, 44(20):10
[6] Robert E. Buckle M. Peter B. Organic Syntheses(Youjihecheng). 1963, 4:722