http://www.chemistrymag.org/cji/1999//011002pe.htm |
Dec.20,
1999 Vol.1 No.1 P.2 Copyright |
Xu Hong, Xu Xianzhong, Li Zonghe
(Department of Chemistry,
Beijing Normal University, Beijing, 100875, China)
Received May 2, 1999; Supported by the National Natural Science Foundation of
China.
*The Chinese version of
this paper was published in Chemistry Online (Huaxue Tongbao), 1999, (13): 62.
Abstract The bond-selective reaction
HOD and H has been studied with the theory of dynamical properties on reaction path (IRC)
in molecular internal coordinates constructed by authors. Compared with the reaction HOH
and H that has no bond selectivity and the reaction H2(u=0,1) and OH that has mode selectivity,
wK (frequencies
orthogonal to IRC) and BKL (coupling constants between breaking bond and the
other vibrations) of every reaction in internal coordinates have been obtained. In order
to reach the object of bond-selective reaction at certain electron state, the theoretic
demands including the vibration frequencies wK of breaking bond and the coupling constants BKL
between breaking bond and the vibrations of other bond (bond angle) in the molecules have
been obtained, based on the analysis of bond selectivity on each reaction.
Keywords bond-selective reaction, molecular internal coordinates, HOD.
Changing productive rate, controlling the
process of chemical reaction and machining molecules at will are goals of chemists. In
many chemical reactions, these goals are partly realized by changing outer conditions
(such as temperature and pressure) or finding the right catalyzer through iterative
experiments. But all of these can only interfere the reaction superficially and can not
reach the goal to clip the molecule following our inclinations, to synthesize the material
we want.
The goal of bond-selective reaction is to machine molecule selectively.
Vibration excited mode is the only way of molecular dissociation and many chemical
reactions. To reach the goal of bond-selective reaction, it is necessary to study the
molecular vibrational excited mode. For the life of local-mode vibrational eigenstates is
too short to observe easily, it results in difficulties in the progress of bond-selective
reaction.
The reaction between OH and vibrationally excited H2, OH+H2(u=1)![]() H2O+H, was studied experimentally at 298 K by R. Zellner
and W. Steinert in 1981[1]. They found that the rate constant for the reaction
was enhanced a factor of 2 when molecule H2 excited to vibrational mode v=1. A
comparison with the rate constant k (u=0) for the ground-state reaction of reaction OH+H2(u=1)
H2O+H, was studied experimentally at 298 K by R. Zellner
and W. Steinert in 1981[1]. They found that the rate constant for the reaction
was enhanced a factor of 2 when molecule H2 excited to vibrational mode v=1. A
comparison with the rate constant k (u=0) for the ground-state reaction of reaction OH+H2(u=1)![]() H2O+H R(1) leads to
H2O+H R(1) leads to
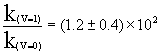
It shows that the reaction has mode selectivity.
In 90's, Zare et al. studied the reaction of HOD and H experimentally[2,3].
The reaction has two main passages: one is HOD+H![]() HO+DH R(2.a),the other is DOH+H
HO+DH R(2.a),the other is DOH+H![]() DO+HH R(2.b). They found that
the OD:OH ratio is 1.38+0.14 for the reaction H+HOD(0,0,0)
(ground-state HOD). In contrast, this ratio is greater than 25:1 (enhanced the product
branching ratios OD/OH more than 18 times)for H+HOD(0,0,1) and less than 1:5.8 (enhanced
the product branching ratios OH/OD more than 8 times) for H+HOD(1,0,0) . Here (0,0,1) and
(1,0,0) denote the O-H and O-D stretch fundamentals respectively. In 1993, Crim et al.
studied the reaction of HOD and H in high excited state experimentally[4]. The
experiment demonstrated that excitation of the third O-H stretching overtone, 4uOH, favors breaking the O-H
bond with product branching ratio OD:OH>100 and excitation of the fourth O-D stretching
overtone, 5uOD,
favors breaking the O-D bond with main product OH. All of these indicated that the
reaction for HOD and H has bond selectivity.
DO+HH R(2.b). They found that
the OD:OH ratio is 1.38+0.14 for the reaction H+HOD(0,0,0)
(ground-state HOD). In contrast, this ratio is greater than 25:1 (enhanced the product
branching ratios OD/OH more than 18 times)for H+HOD(0,0,1) and less than 1:5.8 (enhanced
the product branching ratios OH/OD more than 8 times) for H+HOD(1,0,0) . Here (0,0,1) and
(1,0,0) denote the O-H and O-D stretch fundamentals respectively. In 1993, Crim et al.
studied the reaction of HOD and H in high excited state experimentally[4]. The
experiment demonstrated that excitation of the third O-H stretching overtone, 4uOH, favors breaking the O-H
bond with product branching ratio OD:OH>100 and excitation of the fourth O-D stretching
overtone, 5uOD,
favors breaking the O-D bond with main product OH. All of these indicated that the
reaction for HOD and H has bond selectivity.
A quasiclassical trajectory study of the state-to-state dynamics of H+H2O ![]() OH+H2 had been done by George C.Schatz et al.
in 1984[5]. The work include a detailed study of reagent normal-mode and
local-mode excitation effects, and of product state energy partitioning. Their conclusion
is that if energy transfer between vibrational modes and between vibration and rotation
were neglected, the reaction rate constant of the bond breaking enhanced rapidly when
certain of the local-mode is excited. In 1992, D.C.Clary et al. reported the calculations
of vibrationally state-selective cross section for the H+HOD
OH+H2 had been done by George C.Schatz et al.
in 1984[5]. The work include a detailed study of reagent normal-mode and
local-mode excitation effects, and of product state energy partitioning. Their conclusion
is that if energy transfer between vibrational modes and between vibration and rotation
were neglected, the reaction rate constant of the bond breaking enhanced rapidly when
certain of the local-mode is excited. In 1992, D.C.Clary et al. reported the calculations
of vibrationally state-selective cross section for the H+HOD ![]() H2+OD
reaction with a quantum-mechanical method[6]. Desheng Wang et al. presented
approximate quantum calculations of cumulative reaction probabilities, rotationally
averaged cross sections, and branching ratios for the reactions H+H2O
H2+OD
reaction with a quantum-mechanical method[6]. Desheng Wang et al. presented
approximate quantum calculations of cumulative reaction probabilities, rotationally
averaged cross sections, and branching ratios for the reactions H+H2O![]() OH+H2 and
OH+H2 and ![]() using the
Walch-Dunning-Schatz-Elgersma potential[7].
using the
Walch-Dunning-Schatz-Elgersma potential[7].
The mass-weighted Cartesians coordinates are used to deal with
vibrations in all of the calculations above. Normal-modes of any point along reaction path
are expressed in mass-weighted Cartesians coordinates. The physical diagrams of normal
modes of polyatomic molecules reaction in mass-weighted Cartesians coordinates are
complex, not clear and not easy to understand, which brings limitations to discuss
bond-selective reaction. In the calculations above, all of these chemists did not consider
the reassignment of excited energy and did not compare the results with experimental data
actual. It can not illustrate the situations about transfer of excited bond's energy
between vibrational modes in reaction progress.
To illustrate bond-selective reaction, a set of reaction path theory in
internal coordinates is constructed by authors[8]. We'll use the theory to
discuss the selectivity of reaction HOD with H atom.
1 THE THEORY OF REACTION PATH IN MOLECULAR INTERNAL
COORDINATES
In 1970, Fukui[9] defined that the intrinsic reaction coordinates (IRC) is
a spatial curve (R(s)) along the perpendicular line of the potential face's tangential
plane which connects the points of reactants, transitional state and products. The
reaction coordinates got by this method have no relation with the selection of coordinate
systems. It has the properties of short ground return[10] which is constant in
the change of internal coordinates.
The IRC equation in internal coordinates can be expressed as[11]:
![]()
where g denotes the gradient vector with ![]() ,
,
R is station vector in internal coordinates, W is station energy.G is represented
with elements:
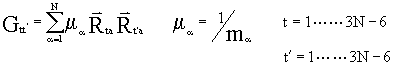 .
.
The
relation between |
and |
|
, where |
is atom a's displacement vector. |
The physical meaning of
the vector ![]() is as follows: Keep all
atoms except atom a in their equilibrium positions.
is as follows: Keep all
atoms except atom a in their equilibrium positions. ![]() is in the direction along which a given
displacement of atom a will produce the greatest increase of
is in the direction along which a given
displacement of atom a will produce the greatest increase of ![]() . The magnitude of
. The magnitude of ![]() is equal to the increase in
is equal to the increase in ![]() produced by unit displacement of the atom
in this most effective direction.
produced by unit displacement of the atom
in this most effective direction.
N is a suitable normalization factor; it can be written as:![]() ,
,
According to IRC equation, the components of motion vector along IRC (LF)
in internal coordinates are determined as:
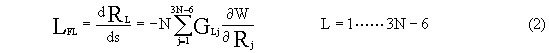 ,
,
where RL is No L internal coordinate of the system, W is the potential of the
system, s is intrinsic reaction coordinate.
From the IRC equation in molecular internal coordinates, vibrational
analysis of the various points along IRC can be done and the energy gradient and force
constant matrix can be obtained. To get the vibrational mode orthogonal to IRC, we define
operator P:
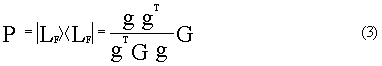 ,
,
where P is a 3N-6 dimensional phalanx.
We define the operator ![]() as:
as:
![]() . I is a 3N-6 dimension unit matrix.
. I is a 3N-6 dimension unit matrix.
When ![]() is used to act on
force constant matrix in internal coordinates the projected force constant matrix is got:
is used to act on
force constant matrix in internal coordinates the projected force constant matrix is got:
![]() (4),
(4),
With GF equation, it can be obtained[12]:
![]() .
.
Solving the equation (5) can get the vibrational frequency w K and vibrational vector ![]() orthogonal to IRC in internal coordinates.
orthogonal to IRC in internal coordinates.
In terms of differential geometry principles, major perpendicular
vector ![]() of IRC curve R(s) defined by Fukui is
the differential vector of R(s)'s tangential line, which is the differential vector of the
motion vector along IRC:
of IRC curve R(s) defined by Fukui is
the differential vector of R(s)'s tangential line, which is the differential vector of the
motion vector along IRC:
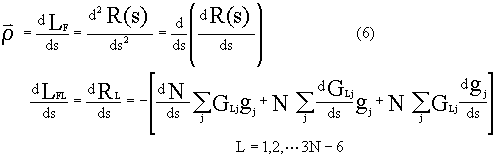 ,
,here
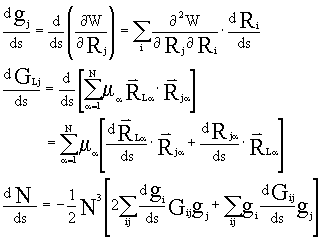
The curvature of IRC is:![]() .
.
So the projected curvature BKF of each vibrational mode LK
in internal coordinates can be easily expressed as:
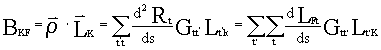 .
(7)
.
(7)
Apparently, BKF can also be regarded as the coupling
constant between the motion vector along IRC and No K vibrational vector orthogonal to IRC
in internal coordinates.Similarly, BKL is defined as the coupling constant
between each two vibrational vectors orthogonal to IRC:![]() in which,
in which, ![]() K,L=1,2,...,3N-7.
The finite difference approximation is applied to the solving of the equation of BKL.
Finally, we obtained:
K,L=1,2,...,3N-7.
The finite difference approximation is applied to the solving of the equation of BKL.
Finally, we obtained:
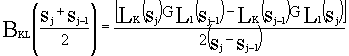 (8)
(8)
So far ,the expressions of all dynamical properties (LF,LK,w K,BKF,BKL) on reaction path in internal coordinates have all
been obtained, and we will use them to study the bond-selective reaction.
2 CALCULATIONS AND DISCUSSION
To study the bond-selective reaction ![]() R(2), we studied the dynamical
properties of typical mode-selective reaction OH+H2(u=0,1)
R(2), we studied the dynamical
properties of typical mode-selective reaction OH+H2(u=0,1) ![]() HOH+H R(1) and the reaction has no bond selectivity H2O+H
HOH+H R(1) and the reaction has no bond selectivity H2O+H ![]() HO+HH R(3) in molecular internal
coordinates at the same time. The molecular internal coordinate system of these reactions
is defined as figure 1:
HO+HH R(3) in molecular internal
coordinates at the same time. The molecular internal coordinate system of these reactions
is defined as figure 1: 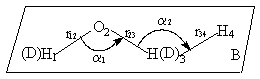
Fig.1 The molecular internal coordinate system of three
reactions R(1),R(2) and R(3)
where r12, r23, r34 are bond lengths, a1, a2 are bond angles, B is dihedral angle (planes 123-234). The molecular internal coordinates are:
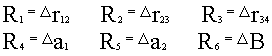
where R1,R2,R3 are internal coordinates of the unreacted bond, the
breaking bond and the forming bond respectively, R4,R5 are internal
coordinates of bond angles in reaction and R6 is internal coordinate of
dihedral angle.
Based on BERNY energy gradient method (at UHF/6-31G** level), the
geometries of reactants, transition state and products in the reaction were optimized, and
the vibrational analysis of the transition state has been done, using computer programs
Gaussian 94.Then starting off from the transition state with GAMESS, based on MOROKUMA
numerical analysis method, the intrinsic reaction coordinates (IRC) is obtained. Later,
according to the IRC equation based on the theory of reaction path in internal
coordinates, LF (motion vector along IRC), P (projection operator), FP
(projected force constant matrix) can all be gained. Both vibrational vectors and
vibrational frequencies orthogonal to IRC in internal coordinates are obtained by solving
the eigenequation ![]() . The coupling constants BKL
are calculated by using the equation (8).
. The coupling constants BKL
are calculated by using the equation (8).
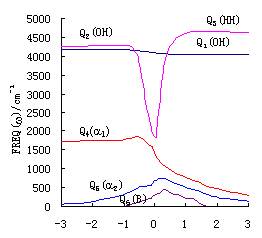
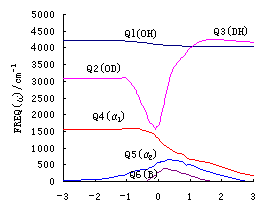
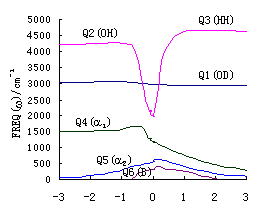
Fig. 2(a),(b),(c) give the change of harmonic
frequencies along reaction coordinates in molecular internal coordinates of the three
reactions R(3\1) ,R(2.a) ,R(2.b). Here Q1 is the change
of stretch vibration of unreacted bond (OH or OD); Q2 is the change of stretch
vibration of reacting bond (OH or OD); Q3 is the change of stretch vibration of
forming bond (HH or DH); Q4 is the change of bend vibration of ![]()
![]() 1(
1(![]() HOH or
HOH or ![]() HOD);Q5
is the change of bend vibration of
HOD);Q5
is the change of bend vibration of![]()
![]() 2(
2(![]() OH(D)H); Q6 is the
change of out-of-plane (B) vibration of the reaction system..
OH(D)H); Q6 is the
change of out-of-plane (B) vibration of the reaction system..
Figure 2 shows that the
frequencies of vibrational mode Q1 change little in these reactions. It
indicates that Q1 hardly take part in reactions. The frequencies of Q2
and Q3 change greatly in the process of the reaction. It reflects the breaking
process of the reacting bond and the forming process of the bond HH(D). The change of Q4
shows the disappearing process of ![]()
![]() 1(
1(![]() HOH or
HOH or ![]() HOD). Q5
and Q6 represent the forming and disappearing process of
HOD). Q5
and Q6 represent the forming and disappearing process of ![]()
![]() 2(
2( ![]() OH(D)H
) and the reaction plane (B) respectively. Apparently all of these are reasonable.
OH(D)H
) and the reaction plane (B) respectively. Apparently all of these are reasonable.
Seen from reverse direction, the Figure 2 (a) shows the change of
vibrational mode of reaction OH+H2 ![]() HOH+H. It is reasonable apparently too.
HOH+H. It is reasonable apparently too.
Observing Figure 2 . (a),(b),(c)
we can find that the obvious difference among the bond-selective reactions R(2) and R(3)
with no bond selectivity is the margin between vibrational frequencies of unreacted bond
and reacting bond of reaction. The margin of R(2.a) and R(2.b) is nearly 1000cm-1,
while the vibrational frequencies of R(3)'s unreacted bond and reacting bond are almost
equal (the margin is only about 100 cm-1). The vibrational frequencies margin
between bond OH and OD is big, thus results in that the respective excited state can be
made easily in the reaction of R(2.a) and R(2.b). However, in the reaction R(3), to excite
one predetermined bond OH by laser is notoriously difficult, it brings difficulties to the
preparation of local-mode high vibrationally excited eigenstates.
|
|
|
Fig.3 (a)
Plots of coupling constants between vibrational mode of breaking bond orthogonal to IRC
and others vibrational modes BKL
vs. reaction coordinate s of reaction R(3\1) (HOH+H
(b) Plots of coupling constants between vibrational mode of breaking bond orthogonal to IRC and others vibrational modes BKL vs. reaction coordinate s of reaction R(2.a)(HOD+H (c) Plots of coupling constants between vibrational mode of breaking bond orthogonal to IRC and others vibrational modes BKL vs. reaction coordinate s of reaction R(2.b)(DOH+H |
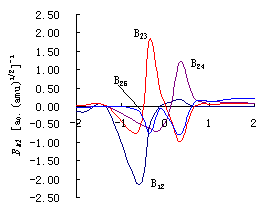
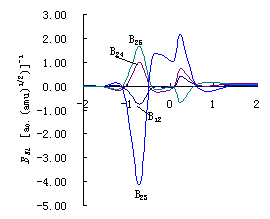
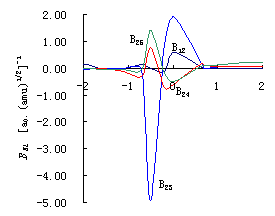
Figure 3. (a),(b),(c) give the mutative curve of coupling constants BKL
between breaking bond and the inner molecular bond (bond angle) or the breaking bond
and intermolecular (interatomic) bond (bond angle) of the reaction in molecular internal
coordinates. Here B12 is the coupling between internal coordinates R1 and R2,
representing unreacted bond (D)H1O2's stretch vibration and reacting
bond O2H(D)3's stretch vibration respectively; B23 is
the coupling constants between internal coordinates R2 and R3, representing the stretch
vibration of the breaking bond O2H(D)3 and the stretch vibration of
forming bond H(D)3H4; B24 is the coupling constants
between internal coordinates R2 and R4, representing the stretch vibration of breaking
bond O2H(D)3 and the bend vibration of bond angle ![]() 1; B25 is the coupling constants between
internal coordinates R2 and R5, representing the stretch vibration of breaking bond O2H(D)3
and the bend vibration of bond angle
1; B25 is the coupling constants between
internal coordinates R2 and R5, representing the stretch vibration of breaking bond O2H(D)3
and the bend vibration of bond angle ![]() 2. The
coupling constants B12 and B24 are inner molecular coupling
constants and B23,B25 are intermolecular coupling constants.
2. The
coupling constants B12 and B24 are inner molecular coupling
constants and B23,B25 are intermolecular coupling constants.
In these two kinds of coupling, the greater the coupling between
breaking bond and inner molecular bond (bond angle) is, the more it does harm to the
reaction; the greater the coupling between breaking bond and intermolecular (interatomic)
bond (bond angle) of the reaction is, the more it is conducive to the process of the
reaction. The exist of the inner molecular coupling results in that the excited energy
transfer to other bonds, not retaining on the excited bond solely, especially when laser
is used to excite the breaking bond. The greater the coupling is, the more quickly the
energy transfer.
Fig.3 (a),(b),(c) show that reactions R(2.a) ,R(2.b) and R(3) all have the coupling
between breaking bond and inner molecular bond (bond angle), but there is no coupling
between breaking bond H-H (which is the molecule itself) and other inner molecular bond
(bond angle) in reaction HO+HH ![]() HOH+H
R(1). The reaction rate will be enhanced 100 times if we excite H2 to one
quantum of vibration u=1 in
reaction R(1)[1], but the reaction R(2) can not be enhanced equally. The
product branching ratio of R(2) is enhanced no more than 18 times[2,3]. In the
local-mode reaction at high excited vibrational eigenstates, the exist of the coupling
between inner molecular vibrational modes made the action of energy transfer easier.
Consequently, the product branching ratio can not be enhanced more than one hundred times
by excitation of OH into 4 quanta of vibration u=4OH or excitation of OD into 5 quanta of vibration u=5OD[4].
HOH+H
R(1). The reaction rate will be enhanced 100 times if we excite H2 to one
quantum of vibration u=1 in
reaction R(1)[1], but the reaction R(2) can not be enhanced equally. The
product branching ratio of R(2) is enhanced no more than 18 times[2,3]. In the
local-mode reaction at high excited vibrational eigenstates, the exist of the coupling
between inner molecular vibrational modes made the action of energy transfer easier.
Consequently, the product branching ratio can not be enhanced more than one hundred times
by excitation of OH into 4 quanta of vibration u=4OH or excitation of OD into 5 quanta of vibration u=5OD[4].
The inner molecular coupling divided into two kinds: one is the
coupling between breaking bond and inner molecular bond angle such as B24, the
other is the coupling between breaking bond and unreacted bond such as B12.
Classic transition state theory indicates that the reaction will go on to the products
once the transition state is overcome. It can be seen clearly from Fig.3 that B24
are almost same before transition states of the former three reactions, but B12
of them are different greatly. The maximum absolute value of coupling constant B12
is more than 2.00 in the reaction HOH+H ![]() HO+HH
R(3), no more than 0.78 in the reaction HOD+H
HO+HH
R(3), no more than 0.78 in the reaction HOD+H ![]() HO+DH R(2.a), is only 0.58 in
the reaction DOH+H
HO+DH R(2.a), is only 0.58 in
the reaction DOH+H ![]() DO+HH R(2.b). According to
these, the order of coupling constants that affect bond selectivity of three reactions is
B12 R(3) > B12 R(2.a) >B12 R(2.b). It can be
deduced that energy transfers quickly to bond H1O2 when bond O2H3
is excited in reaction R(3). Consequently, the energy of reacting bond O2H3
fall down, the reaction has no bond selectivity. When bond OD or bond OH of HOD is
excited, the energy transfers slowly for the coupling is little in reaction R(2). So the
reaction has bond selectivity. These are consistent with the experimentally qualitative
analysis of Bronikowski[2,3] and Zellner[4].
DO+HH R(2.b). According to
these, the order of coupling constants that affect bond selectivity of three reactions is
B12 R(3) > B12 R(2.a) >B12 R(2.b). It can be
deduced that energy transfers quickly to bond H1O2 when bond O2H3
is excited in reaction R(3). Consequently, the energy of reacting bond O2H3
fall down, the reaction has no bond selectivity. When bond OD or bond OH of HOD is
excited, the energy transfers slowly for the coupling is little in reaction R(2). So the
reaction has bond selectivity. These are consistent with the experimentally qualitative
analysis of Bronikowski[2,3] and Zellner[4].
3 CONCLUSION
The bond-selective reaction of HOD with H atom has been discussed in this article with
the theory of dynamical properties on reaction path (IRC) in molecular internal
coordinates constructed by authors. It indicates that in order to break a predetermined
bond (bond selective reaction) between an atom or atomic group of certain molecular on one
end of predetermined bond and another molecule, atomic or atomic group, to reach the goal
of bond-selective by excitation of the chosen bond with laser, these are required on
theory:
(1) The bigger the margin between the predetermined bond's vibrational frequency of
certain molecule and the ambient bond's (bond angle's) vibrational frequency is, the more
benefic it is to the preparation of the bond's local-mode vibrationally excited
eigenstates exactly. The margin would be better more than 1000 cm-1.
(2) The smaller the coupling between the predetermined bond and the other inner molecular
bond (bond angle) of the molecule is, the more selective the reaction is, especially the
coupling between the predetermined bond and the other inner molecular bond. It would be
better that the coupling constant is less than 0.7.
REFERENCES
[1] Zellner R, Steinert W. Chem. Phys. Lett., 1981, 81 (3): 568.
[2] Michael J B, William R S, Richard N Z. J. Chem. Phys., 1991, 95 (11): 8647.
[3] Michael J B, William R S, Richard N Z. J. Phys. Chem., 1993, 97: 2194.
[4] Ricardo B M, John D T, Joann M P, Fleming F C. J. Chem. Phys., 1993, 99 (3): 1744.
[5] George C S, Mitchell C C, John L G. J. Phys. Chem., 1984, 88: 2971.
[6] Clary D C. Chem. Phys. Lett., 1992, 192: 34.
[7] Deshen W, Joel M B. J. Chem. Phys., 1993, 98 (8): 6235.
[8] Cao X Y, Li Z H, Zhang Y J et al. Chemistry of Hongkong, 1997, (2): 29.
[9] Fukui K. J. Phys. Chem., 1970, 74 (23): 4161.
[10] Sana M, Reckinger G, Leroy G. Theoret. Chim. Acta., 1981, 58: 145.
[11] Gonzalez C, Schlegel H B. J. Phys. Chem., 1990, 94: 5523.
[12] Wilson E B, Decius J C, Cross P C. The theory of infrared and Raman
vibrational spectra. Dover, 1980.