http://www.chemistrymag.org/cji/2000/02a045pe.htm |
Oct. 1, 2000 Vol.2 No.10 P.45 Copyright |
Ye Qingguo, Zhong Chongli #
(Dept. of Chem. Eng., Qingdao Institute of Chemical Technology, Qingdao 266042; #Dept.
of Chem. Eng., Beijing University of Chemical Technology, Beijing 100029, China)
Received Jun. 11, 2000.
Abstract The combinatorial part of the
original UNIFAC model is modified to improve its predictive accuracy for asymmetric
systems by replacing the volume parameter with an "effective" one. A relation is derived between the "effective" and the original volume parameters. The improved UNIFAC model,
the newly proposed combinatorial part coupled with the original residual part, woks well
for asymmetric systems such as polymer solutions and those containing small and large
molecule than the original UNIFAC model, which is also workable for systems containing
only normal fluids. Comparing to those UNIFAC-FV models, the new model has the advantage
of not requiring additional information, especially liquid molar volumes, in the
calculation, which is very convenient for practical use. Parallel calculations are also
carried out for another recently modified UNIFAC model as a comparison.
Keywords Prediction, Activity coefficient, UNIFAC model, Group contribution,
Asymmetric system.
1 INTRODUCTION
The UNIFAC model [1] is widely used for phase equilibrium calculations. For
normal fluid mixtures, its predictive accuracy is usually good enough to meet the
engineering demands, however, when highly asymmetric systems are dealt with, such as
polymer solutions, it generally performs poorly. To improve it for such systems, Oishi and
Prausnitz [2] added a free volume (FV) term to develop the so-called UNIFAC-FV
model, which gives greatly improved predictions for solvent activities in polymer
solutions. Some modified versions have been developed [3,4], however, similar
to the UNIFAC-FV model of Oishi and Prausnitz[2], they all require pure
component liquid molar volumes in the calculation, which is troublesome for systems where
such volumes are not accurately known, especially for those containing supercritical
fluids. Consequently, an alternative method for extending the original UNIFAC model to
highly asymmetric systems was proposed whose main idea is not to introduce liquid molar
volumes in the modified ones. We, here, call this kind models as liquid volume free (LVF)
modified UNIFAC models. Two such models have been developed, one is proposed by Voutsas et
al. [5], the other one is proposed by us [6]. The previous
investigation [5,6] show that such models work as well as or even better than
those UNIFAC-FV type ones [2-4] for prediction of activity coefficients of
highly asymmetric systems, however, the LVF ones are more convenient for engineering use.
In this work our previously modified UNIFAC model [6] is
further improved to make it able to deal with both asymmetric and symmetric systems. A
large number of experimental data, including polymer/solvent, small/large molecule and
normal/normal fluid systems, were used to test the new model and compared with that
proposed by Voutsas et al.[5] which has not been tested extensively before.
2 THE PROPOSED
COMBINATORIAL ACTIVITY COEFFICIENT MODEL
2.1 The proposed combinatorial activity coefficient model
The activity coefficient for component i form the original UNIFAC model [1] is
as follows:
ln gi = ln giC + ln giR
(1)
Where giC
and giR
are the combinatorial and the residual parts, respectively.
To improve it for polymer solutions, in our previous work[6]
the residual part was remained unchanged while the combinatorial part was modified as
follows :
ln giC = ln(fi'/xi)+1-(fi'/xi)-0.5zqi [ln(fi/qi)+1-(fi/qi)]
(2)
where fi = xiri/Sxjrj
(3)
qi = xiqi/Sxjqj
(4)
and fi'= xiri'/Sxjrj'
(5)
with ri'=ri,
for small molecule
(6)
ri'=(0.6583+0.3709/n)ri , for
polymer (7)
=0.6583ri , for polymer
(8)
where xi,
ri and qi are
mole fraction, volume and surface area parameters for component i, respectively. n is the
number of monomers constituting the polymer. ri' is
the proposed "effective"
volume parameter. Eq.(2) is firstly proposed by Kikic et al.[7], which reduces
to the expression of the original UNIFAC model[1] if fi' is set to be identical to fi.
Obviously, the key point of our modified
combinatorial activity coefficient model is an “effective” volume parameter, ri', is
used in the calculation of fi', and
a relation between the "effective"
volume parameter ri' and the real ri
for polymer was proposed, which stemmed from the theoretical approximate relation between
the excluded volume of an n-mer chain molecule and that of the monomer [6]. It
is evident that our modified UNIFAC model does not need liquid molar volumes in the
calculation for polymer/solvent systems, which requires just the same information as that
by the original UNIFAC model.
Though an explicit free volume term is not included in our modified
UNIFAC model, the use of an "effective"
volume parameter for polymer does a good contribution to the free volume effects. Our
previous work shows that the new modified UNIFAC model works better than the UNIFAC-FV
model [2] for prediction of solvent activities in polymer solutions. As only
polymer/solvent systems were dealt in our previous work, the approximate equation ri'=0.6583ri was adopted since n is large for a
polymer. In this work, we want to extend the model to both asymmetric and symmetric
systems containing small, large as well as polydisperse molecules. As a result, eq.(7) is
remained. Since the coefficient in eq.(7) is not unit when n is set to one, which is
caused by the use of an approximate relation between the excluded volume for an n-mer
chain molecule and that for the monomer, eq.(7) was adjusted a little as follows:
ri'=(0.6583+0.3417/n)ri , for
polymer or large molecule (9)
Furthermore, n is redefined as follows so that eq.(9)can be used for
systems containing both large and small molecules:
n = VvdW,large/VvdW,small = rlarge/rsmall
(10)
where VvdW is the van der Waals
volume. Obviously, eq.(10) means that the "monomer" constituting the large molecule has the same vdW volume as the
smaller one for a binary system. Eqs.(1)-(6),(9) and (10) constitute the modified UNIFAC
model proposed in this work which is denoted as the UNIFAC-r model for convenience.
Obviously, eq.(9) reduces to our previous expression, that is eq.(8),
for polymer/solvent systems, and for systems with equal molecular sizes, the UNIFAC-r
model reduces exactly to the original UNIFAC model. Therefore, it is expected that the
proposed UNIFAC-r model can be used for both asymmetric and symmetric systems.
2.2 The R-UNIFAC model[5]
Another liquid molar volume free combinatorial activity coefficient model applicable to
asymmetric systems is the so-called R-UNIFAC model proposed by Voutsas et al.[5],
where eq.(2) is also adopted with a different expression for fi' as
follows:
fi' = xiriR/SxjrjR
(11)
and R = 0.9[1-(VvdW,small/VvdW,large )]=0.9[1- (rsmall/rlarge)]
(12)
From their investigation the R-UNIFAC
model works as well as or even better than those free volume ones [5] for
athermal asymmetric mixtures. We will test and compare systematically the two liquid molar
volume free modified UNIFAC models in this work. As only the combinatorial activity
coefficient model is given in the work of Voutsas et al. [5], the residual part
of the original UNIFAC model [1] is used, the same as done in our model, as the
residual one for the R-UNIFAC model to constitute a complete model applicable to both
athermal and thermal mixtures.
3 RESULTS AND DISCUSSION
3.1 Prediction of solvent activities in polymer solutions
Vapor-liquid equilibrium of polymer solutions are important in the processing of many
polymeric materials. As the original UNIFAC model [1] does not work well for
these systems, many efforts, as mentioned above, have been made to improve it. Our
modified UNIFAC model, the UNIFAC-r model, is proposed mainly for this purpose with the
advantage of not requiring liquid molar volumes in the calculation.
In our precious work [6], a total of 51 data sets for
polymer solutions were adopted to test the proposed model, 36 additional data sets were
added in this work. As a result, a large data bank including 87 data sets is available to
test the new model and compare with the original UNIFAC as well as the R-UNIFAC models.
The predicted results, that is, the average absolute deviation (AAD) of solvent
activities, were listed in Table 1. Obviously, the original UNIFAC model gives large AADs,
usually about 20%, for most systems, while the UNIFAC-r model shows greatly improved
predictions with the AADs within 10% for most systems. The R-UNIFAC model shows improved
predictions for many systems, however, it performs fairly poor for some ones. Comparing
the two modified UNIFAC models, it is evident that the UNIFAC-r model works better that
the R-UNIFAC model for polymer solutions.
System* |
Temp. |
No. of |
AADa**,% |
Ref. |
||
PVA(1.11 ×105)+ethyl acetate |
303 |
6 |
21.8 |
6.5 |
4.8 |
[8] |
PVA(8.6 ×103)+ethyl acetate |
303 |
8 |
23.2 |
5.0 |
7.7 |
[8] |
PVA(1 ×106)+acetone |
303 |
4 |
21.4 |
11.0 |
6.2 |
[8] |
PVA(1.1 ×105)+acetone |
303 |
6 |
20.7 |
6.0 |
5.3 |
[8] |
PVA(8.6 ×103)+acetone |
303 |
5 |
21.0 |
1.3 |
4.7 |
[8] |
PVA(1.1 ×105)+ benzene |
303 |
11 |
17.7 |
7.0 |
2.8 |
[8] |
PVA(4.8 ×104)+benzene |
303 |
7 |
12.9 |
11.2 |
3.8 |
[9] |
PVA(1.4 ×105)+benzene |
303 |
8 |
20.1 |
13.7 |
7.9 |
[9] |
PVA(1.1 ×105)+methanol |
303 |
9 |
21.1 |
41.6 |
10.3 |
[8] |
PS(2.2 ×105)+cyclohexane |
313-353 |
28 |
25.5 |
11.5 |
6.0 |
[8] |
PS(1 ×105)+cyclohexane |
303-333 |
19 |
21.0 |
6.3 |
5.8 |
[8] |
PS(6.3 ×104)+benzene |
288-333 |
31 |
15.8 |
3.7 |
3.2 |
[9] |
PS(5 ×105)+benzene |
293 |
15 |
23.7 |
14.4 |
7.2 |
[9] |
PS(9 ×105)+benzene |
288-333 |
30 |
13.9 |
10.8 |
2.2 |
[9] |
PS(1 ×104)+toluene |
322 |
9 |
20.8 |
1.3 |
2.9 |
[9] |
PS(9 ×105)+toluene |
298 |
5 |
9.1 |
5.8 |
0.9 |
[9] |
PS(2.9 ×105)+toluene |
298-353 |
16 |
17.8 |
10.2 |
2.8 |
[9] |
PS(6 ×105)+toluene |
297 |
5 |
18.4 |
24.2 |
3.3 |
[8] |
PS(5.4 ×104)+m-xylene |
403-448 |
22 |
33.1 |
10.3 |
6.3 |
[9] |
PS(2.9 ×105)+chloroform |
298-323 |
21 |
16.7 |
11.8 |
5.3 |
[9] |
PS(9 ×104)+chloroform |
298-323 |
6 |
17.3 |
5.1 |
4.4 |
[9] |
PS(6 ×105)+dichloromethane |
297 |
6 |
11.8 |
36.7 |
10.7 |
[8] |
PS(6 ×105)+tetrachloromethane |
297 |
4 |
13.4 |
36.8 |
10.4 |
[8] |
PS(5 ×105)+tetrachloromethane |
293 |
14 |
14.5 |
23.1 |
4.3 |
[9] |
PS(2.9 ×105)+propyl acetate |
298-343 |
19 |
16.3 |
16.7 |
2.3 |
[8] |
PS(1.6 ×104)+2-propanone |
298-333 |
15 |
20.0 |
8.8 |
5.8 |
[9] |
PS(2.9 ×105)+2-butanone |
298-343 |
17 |
16.4 |
25.6 |
5.1 |
[9] |
PS(1 ×104)+2-butanone |
322 |
9 |
13.3 |
10.6 |
5.7 |
[9] |
PS(2 ×105)+3-pentanone |
293 |
12 |
32.9 |
13.0 |
16.9 |
[9] |
PS(5 ×105)+3-pentanone |
293 |
11 |
33.5 |
7.8 |
18.7 |
[9] |
PS(5.4 ×104)+ n-nonane |
403-448 |
16 |
31.2 |
14.5 |
3.4 |
[9] |
PIB(4 ×104)+ n-pentane |
298-328 |
22 |
22.1 |
2.8 |
9.1 |
[8] |
PIB(2.3 ×106)+ n-pentane |
298-328 |
33 |
15.8 |
1.3 |
8.7 |
[9] |
PIB(5 ×104)+ n-hexane |
298-338 |
23 |
31.9 |
3.2 |
11.9 |
[10] |
PIB(4 ×104)+ n-octane |
298 |
5 |
4.8 |
1.2 |
2.1 |
[11] |
PIB(4.7 ×106)+ cyclopentane |
297 |
6 |
30.3 |
42.8 |
5.5 |
[8] |
PIB(4 ×104)+ cyclohexane |
298 |
8 |
24.1 |
1.0 |
7.8 |
[12] |
PIB(1 ×105)+ cyclohexane |
298-338 |
30 |
16.0 |
1.9 |
5.6 |
[9] |
PIB(4 ×104)+ benzene |
298-314 |
25 |
21.6 |
4.4 |
7.1 |
[9] |
PIB(4.5 ×104)+ benzene |
298-338 |
29 |
18.9 |
5.9 |
4.4 |
[9] |
PIB(7.6 ×104)+ benzene |
300 |
22 |
25.6 |
6.9 |
7.1 |
[9] |
PIB(5 ×104)+ toluene |
298-338 |
23 |
33.4 |
13.2 |
12.0 |
[10] |
PIB(5 ×104)+ ethyl benzene |
298-338 |
20 |
20.6 |
17.2 |
9.1 |
[10] |
PIB(4.7 ×106)+2,2-dimethyl butane |
297 |
4 |
37.7 |
14.3 |
18.3 |
[8] |
| PB(6.5×104)+ n-hexane | 297 |
4 |
23.0 |
18.8 |
2.6 |
[13] |
PB(6.5 ×104)+ cyclohexane |
297 |
4 |
14.7 |
23.9 |
9.3 |
[13] |
PB(4 ×104)+ benzene |
300 |
8 |
6.6 |
24.2 |
13.3 |
[9] |
PB(6.5 ×104)+ toluene |
297 |
5 |
15.6 |
23.4 |
8.4 |
[13] |
PB(2.5 ×105)+ ethyl benzene |
353-403 |
35 |
24.9 |
31.7 |
9.5 |
[9] |
PB(2.5 ×105)+ n-nonane |
353-403 |
33 |
35.3 |
18.1 |
13.0 |
[9] |
PEO(6 ×105)+ benzene |
323-343 |
13 |
6.2 |
17.1 |
5.8 |
[14] |
PEO(5.7 ×103)+ benzene |
319-346 |
14 |
8.2 |
7.3 |
5.5 |
[14] |
PEO(1.5 ×103)+ water |
298-308 |
24 |
6.1 |
8.5 |
8.7 |
[9] |
PEO(4.2 ×103)+ water |
298-308 |
24 |
7.6 |
11.1 |
11.9 |
[9] |
PPO(5 ×105)+ benzene |
320-348 |
26 |
7.2 |
19.7 |
6.6 |
[15] |
PPO(3.4 ×103)+ methanol |
248-298 |
20 |
1.3 |
1.3 |
1.0 |
[9] |
PPO(1.0 ×103)+methanol |
248-298 |
20 |
3.2 |
5.3 |
5.0 |
[9] |
PPO(2.0 ×103)+methanol |
248-298 |
20 |
1.6 |
1.7 |
1.4 |
[9] |
POD(2.2 ×105)+toluene |
303 |
13 |
26.4 |
10.9 |
14.9 |
[16] |
POD(1.9 ×105)+toluene |
303 |
6 |
29.3 |
6.0 |
16.0 |
[9] |
PD(2.1 ×105)+toluene |
303 |
17 |
10.9 |
37.7 |
12.8 |
[16] |
PDD(9.5 ×104)+toluene |
303 |
20 |
20.8 |
12.2 |
6.2 |
[16] |
PH(2.2 ×105)+toluene |
303 |
21 |
13.9 |
30.7 |
10.2 |
[16] |
PMA(6.3 ×104)+benzene |
297 |
7 |
19.6 |
14.5 |
2.2 |
[17] |
PMA(6.3 ×104)+toluene |
297 |
5 |
20.4 |
19.3 |
5.1 |
[17] |
PEA(3.4 ×104)+benzene |
297 |
6 |
16.0 |
10.3 |
2.6 |
[17] |
PEA(3.4 ×104)+toluene |
297 |
6 |
21.3 |
8.8 |
3.7 |
[17] |
PBA(3.3 ×104)+benzene |
297 |
7 |
12.5 |
14.8 |
6.6 |
[17] |
PBA(3.3 ×104)+toluene |
297 |
5 |
14.6 |
15.3 |
6.6 |
[17] |
PMMA(3.3 ×104)+chloroform |
297 |
8 |
22.7 |
2.2 |
7.6 |
[17] |
PMMA(3.3 ×104)+dichloromethane |
297 |
7 |
22.6 |
3.5 |
5.2 |
[17] |
PMMA(2.0 ×104)+2-butanone |
321 |
8 |
25.0 |
5.2 |
9.7 |
[9] |
PMMA(2.0 ×104)+toluene |
321 |
8 |
24.5 |
5.5 |
9.0 |
[9] |
PEMA(1.4 ×105)+benzene |
297 |
5 |
16.7 |
16.9 |
2.9 |
[17] |
PEMA(1.4 ×105)+toluene |
297 |
6 |
23.2 |
11.7 |
3.2 |
[17] |
PIP(1 ×105) +tetrachloromethane |
297 |
7 |
14.0 |
28.0 |
10.0 |
[13] |
PIP(4 ×104)+benzene |
283-313 |
23 |
11.8 |
27.2 |
12.9 |
[9] |
PIP(1 ×105)+toluene |
297 |
4 |
12.2 |
30.4 |
12.6 |
[9] |
PIP(1 ×105)+cyclohexane |
297 |
6 |
16.7 |
20.7 |
5.4 |
[13] |
PIP(1 ×105)+2-butanone |
298-318 |
18 |
11.5 |
37.2 |
14.9 |
[9] |
PVME(1.4 ×104)+chloroform |
298 |
13 |
18.5 |
4.7 |
4.7 |
[18] |
PVME(1.5 ×104)+benzene |
323-343 |
13 |
6.1 |
9.0 |
5.9 |
[18] |
PVME(1.5 ×104)+chlorobenzene |
343-373 |
22 |
17.7 |
1.7 |
3.9 |
[18] |
PDMS(4.2 ×103)+benzene |
298-313 |
29 |
12.1 |
19.2 |
15.2 |
[9] |
PP(1.5 ×104)+tetrachloromethane |
298 |
6 |
18.8 |
4.9 |
6.4 |
[9] |
PP(2 ×104)+3-pentanone |
298 |
17 |
28.7 |
13.2 |
14.8 |
[9] |
PVC(3.4 ×104)+toluene |
316 |
8 |
35.7 |
12.4 |
19.1 |
[9] |
PPO: poly(propylene oxide); POD:polyoctadecene; PDD: polydodecene; PD: polydecene; PH polyheptene;
PMA: poly (methyl acrylate); PEA: poly (ethyl acrylate); PBA; poly (n-butyl acrylate); PMMA: poly (methyl methacrylate);
PEMA: poly (ethyl methacrylate); PIP:polyisoprene; PVME: poly(vinyl methyl ether); PDMS: poly(dimethyl siloxane);
PP: polypropylene; PVC: poly (vinyl chloride)
**

3.2 Prediction of activity coefficients for
systems containing a small and a large molecules
In the regression of the original UNIFAC group interaction parameters only the systems
containing normal fluids were included. For those systems containing both small and large
molecules, due to the high asymmetry, and, of course, also partly due to the exclusion in
the regression of the interaction parameters, the original UNIFAC model gives relatively
large errors. In this work, we will test the capability of the proposed UNIFAC-r model for
this kind of systems.
A total of 20 systems were collected as shown in Table 2, where the "asymmetry", that is, the ratio of
the van der Waals volumes of the constituents, were also listed. Though the activity
coefficients for the smaller component are available, those for the larger component are
usually scarce. The predicted results reported in Table 2 show that the proposed UNIFAC-r
model gives better predictions than the original UNIFAC model for activity coefficients of
both components, which are comparable to that from the R-UNIFAC model.
System* |
Temp. |
NP |
r1/r2 | AAD g1**,% AADg2%UNIFAC R-UNIFAC UNIFAC-r UNIFAC R-UNIFAC UNIFAC-r |
Ref. |
|||||
n-C11+n-hexane |
308 |
12 |
1.75 |
4.0 |
0.5 |
1.5 |
3.4 |
0.4 |
1.3 |
[19] |
n-C12+cyclohexane |
288-312 |
32 |
2.11 |
- |
- |
- |
4.9 |
2.1 |
1.1 |
[20] |
n-C12+n-hexane |
308 |
12 |
1.90 |
4.9 |
0.5 |
1.9 |
4.4 |
0.4 |
1.7 |
[21] |
n-C16+benzene |
298-328 |
20 |
3.53 |
3.7 |
6.4 |
2.8 |
4.6 |
10.4 |
5.4 |
[22] |
n-C16 +tetrachloromethane |
298-328 |
22 |
3.32 |
6.2 |
4.7 |
1.7 |
7.4 |
6.0 |
2.1 |
[22] |
n-C16 + 1,2-dichloropropane |
298 |
15 |
3.12 |
16.6 |
4.1 |
8.8 |
12.3 |
3.5 |
6.0 |
[23] |
n-C16 +1,3-dichloropropane |
298 |
12 |
3.12 |
16.6 |
10.0 |
10.5 |
17.8 |
9.1 |
11.8 |
[23] |
n-C16 +cyclohexane |
298-312 |
27 |
2.78 |
- |
- |
- |
4.3 |
3.0 |
0.5 |
[24] |
n-C16 +n-hexane |
298 |
10 |
2.50 |
7.3 |
0.3 |
2.9 |
8.9 |
0.2 |
3.5 |
[25] |
n-C16 +n-octane |
298 |
17 |
1.92 |
3.3 |
0.9 |
0.9 |
4.6 |
1.2 |
1.3 |
[25] |
n-C18 +n-pentane |
303 |
18 |
3.30 |
- |
- |
- |
18.1 |
2.5 |
8.1 |
[26] |
n-C18 +n-hexane |
303 |
16 |
2.80 |
- |
- |
- |
12.1 |
0.3 |
4.6 |
[26] |
n-C18 +2-methyl pentane |
303 |
14 |
2.80 |
- |
- |
- |
11.1 |
1.4 |
5.1 |
[26] |
n-C18 +3-methyl pentane |
303 |
11 |
2.80 |
- |
- |
- |
7.8 |
0.2 |
2.7 |
[26] |
n-C18 +2,2-dimethyl butane |
303 |
13 |
2.80 |
- |
- |
- |
15.0 |
2.1 |
7.0 |
[26] |
n-C18 +2,3-dimethyl butane |
303 |
11 |
2.80 |
- |
- |
- |
9.6 |
0.3 |
3.7 |
[26] |
n-C18 +n-heptane |
303 |
15 |
2.43 |
- |
- |
- |
10.0 |
1.6 |
3.1 |
[26] |
n-C18 +2-methyl hexane |
303 |
10 |
2.43 |
- |
- |
- |
6.5 |
0.4 |
2.2 |
[26] |
n-C18 +2,4-dimethyl pentane |
303 |
12 |
2.43 |
- |
- |
- |
11.8 |
2.6 |
6.3 |
[26] |
n-C20 +cyclohexane |
306-317 |
24 |
3.45 |
- |
- |
- |
7.0 |
6.5 |
1.9 |
[27] |
*
n-C11: n-undecane; n-C12: n-dodecane; n-C16: n-hexadecane; n-C18: n-octadecane; n-C20: n-eicosane**
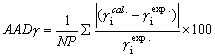
3.3 Prediction of activity coefficients for
systems containing normal fluids
The above calculations show that the proposed UNIFAC-r model gives improved predictions
for highly asymmetric systems where the original UNIFAC model does not work well. It is
necessary to further test if it is also workable for those systems containing only normal
fluids where the original UNIFAC model usually works very well. 22 systems were collected
which were listed in Table 3 according to an increase in the system asymmetry. From the
results obtained, it is clear that the UNIFAC-r model gives similar or even better
predictive accuracy than that of the original UNIFAC model for systems with an asymmetry
smaller than 2.0, which is also the case for the R-UNIFAC models. However, when the
asymmetry is larger than 2.0, both the UNIFAC-r and the R-UNIFAC models, as shown in Table
3, give worse results than of the original UNIFAC model, which is caused by the overlarge
correction of the combinatorial part of the original one. As a result, it is recommended
that the UNIFAC-r model should be used carefully for normal fluid systems with an
asymmetry larger than 2.0, which is also the case for the R-UNIFAC model.
Table 3 Predicted results of activity coefficients for systems containing normal fluids
System |
Temp. |
|
r1/r2 |
AAD g1,% AADg2%UNIFAC R-UNIFAC UNIFAC-r UNIFAC R-UNIFAC UNIFAC-r |
|
|||||
2-propanol + 2-butanone |
328 |
9 |
1.0 |
5.1 |
5.1 |
5.1 |
4.4 |
4.4 |
4.4 |
[28] |
toluene + n-pentane |
293-313 |
45 |
1.03 |
3.1 |
3.1 |
3.1 |
2.6 |
2.6 |
2.6 |
29[] |
chloroform + acetone |
298-308 |
22 |
1.12 |
4.7 |
4.6 |
4.6 |
2.9 |
2.9 |
2.9 |
[30] |
n-hexane + n-butanol |
323-348 |
32 |
1.15 |
5.8 |
5.6 |
5.7 |
8.3 |
8.1 |
8.2 |
[31] |
chlorobenzene + benzene |
298 |
7 |
1.20 |
0.4 |
0.5 |
0.4 |
0.3 |
0.5 |
0.4 |
[32] |
benzene + ethanol |
298 |
9 |
1.24 |
1.5 |
2.0 |
1.8 |
2.0 |
2.4 |
2.2 |
[]33 |
2-butanone + ethanol |
328 |
14 |
1.26 |
4.0 |
5.1 |
4.6 |
2.4 |
3.1 |
2.8 |
[28] |
n-octane + n-hexane |
298 |
10 |
1.30 |
1.0 |
0.1 |
0.4 |
1.0 |
0.1 |
0.5 |
[25] |
n-butanol + 1,2-dichloroethane |
323 |
14 |
1.34 |
3.3 |
4.0 |
3.6 |
1.1 |
2.5 |
1.7 |
[34] |
n-hexane + benzene |
303-333 |
32 |
1.41 |
1.5 |
2.3 |
1.5 |
0.6 |
0.7 |
0.5 |
[29] |
benzene + acetic acid |
298-318 |
32 |
1.45 |
6.1 |
4.4 |
5.1 |
5.4 |
3.8 |
4.5 |
[35] |
n-hexane + 1,2-dichloroethane |
323 |
15 |
1.54 |
3.3 |
2.2 |
2.0 |
2.8 |
1.6 |
2.0 |
[36] |
methanol + water |
323-333 |
52 |
1.56 |
1.2 |
1.6 |
0.9 |
2.7 |
5.2 |
4.1 |
[37] |
n-heptane + benzene |
298-313 |
20 |
1.62 |
7.2 |
3.3 |
5.0 |
2.7 |
1.2 |
1.7 |
[38] |
n-hexane + ethanol |
298 |
9 |
1.75 |
3.3 |
5.3 |
4.1 |
3.8 |
5.4 |
4.3 |
[33] |
n-heptane + 1,2-dochloroethane |
323 |
16 |
1.77 |
2.4 |
2.9 |
1.2 |
2.2 |
2.9 |
1.3 |
[36] |
n-octane + benzene |
298-328 |
18 |
1.83 |
2.0 |
3.4 |
1.6 |
1.1 |
3.2 |
1.5 |
[38] |
cyclohexane + acetic acid |
298-318 |
34 |
1.84 |
6.5 |
5.3 |
5.4 |
6.1 |
5.8 |
5.6 |
[35] |
n-octane+1,2-dichloroethane |
323 |
15 |
2.00 |
4.0 |
10.9 |
7.4 |
1.5 |
6.0 |
3.3 |
[36] |
2-butanone + methanol |
323 |
13 |
2.27 |
1.1 |
6.4 |
3.4 |
2.5 |
6.6 |
2.8 |
[28] |
n-heptane + acetic acid |
298-318 |
26 |
2.35 |
5.0 |
10.9 |
6.9 |
4.8 |
9.8 |
7.1 |
[35] |
ethanol + water |
323-333 |
107 |
2.80 |
1.8 |
6.5 |
3.5 |
2.3 |
15.3 |
9.3 |
[37] |
4 CONCLUSIONS
The improved UNIFAC model, the UNIFAC-r model, proposed in this work is applicable to both
symmetric and asymmetric systems. Its main advantage is that it does not require liquid
molar volumes in the calculation for highly asymmetric systems. The proposed UNIFAC-r
model was tested against various kind of systems including polymer/solvent, small/large
molecule as well as normal/normal fluid systems, and was compared with the original UNIFAC
and the R-UNIFAC models. The results for polymer solutions show that the proposed UNIFAC-r
model gives much better predictions than that of the original UNIFAC model. The R-UNIFAC
model, however, gives good predictions for most systems but performs poorly for some ones.
As to systems containing small and large molecules, the UNIFAC-r model shows comparable
accuracy to the R-UNIFAC model, and both of them work better than the original one.
Furthermore, it is clear that the UNIFAC-r model, as well as the R-UNIFAC model, gives
similar predictions to that of the original UNIFAC model for normal fluid systems when the
system asymmetry is smaller than 2.0. However, they should be avoided using for those
normal fluid systems with a asymmetry larger than 2.0.
Comparing to those UNIFAC-FV type models, the proposed UNIFAC-r model
does not require additional information excepting those required by the original UNIFAC
model in the calculation. Particularly, it does not need liquid molar volumes, so this
UNIFAC-r model is very convenient for engineering use especially for those processes
containing supercritical fluids.
NOMENCLATURE
a
activity
n
ratio of can der waals volumes defined by eq.(10)
NP number of data
points
q
surface area parameter
r
volume parameter
R
parameter
defined by eq.(12)
r'
effective volume parameter
VvdW van der
Waals volume
x
mole fraction
z
coordination number, 10
g
activity coefficient
q
surface area fraction
f', f volume
fraction
Subscripts
i
component i
Superscripts
C
combinatorial part
cal. calculated value
exp. experimental value
R residual part
REFERENCE
[1] Fredenslund A, Jones R L, Prausnitz J M. ALChE J, 1975, 21: 1086.
[2] Oishi T, Prausnitz J M. Ind. Eng. Chem. Process Des. Dev., 1978, 17: 333.
[3] Elbro H S, Fredenslund A, Rasmussen P. Macromolecules, 1990, 12: 4707.
[4] Kontogeorgis G M, Coutsikos P, Tassios D et al. Fluid Phase Equilibria, 1994,
92: 35.
[5] Voutsas E C, Kalospiros N S, Tassios D P. Fluid Phase Equilibria, 1995, 109: 1.
[6] Zhong C, Sato Y, Masuoka H et al. Fluid Phase Equilibria, 1996, 123: 97.
[7] Kikic I, Alessi P, Rasmussen P et al. Can. J. Chem. Eng, 1980, 58: 253
[8] Wohlfarth C. Vapor-Liquid Equilibrium Data of Binary Polymer Solutions. Elsevier,
1994.
[9] Wen H, Elbro H S, Alessi P. Polymer Solution Data Collection Part 1. Vapor-Liquid
Equilibrium; DECHEMA Chemistry Data Series; Frankfurt, 1992.
[10] Masuoka H, Murashige N, Yorizane M. Fluid Phase Equilibria, 1984, 18: 155.
[11] Flory P J, Ellenson J L, Eichinger B E. Macromolecules, 1968, 1: 279.
[12] Eichinger B E, Flory P J. Trans. Faraday Soc., 1968, 64: 2061.
[13] Saeki S, Holste J C, Bonner D C. J. Polym. Sci. Polym. Phys. Ed., 1982, 20: 793.
[14] Booth C, Devoy C J. Polymer, 1971, 12: 309.
[15] Booth C, Devoy C J. Polymer, 1971, 12: 320.
[16] Tait P J T, Livesey P J. Polymer, 1970, 11: 359.
[17] Saeki S, Holste JC, Bonner D C. J. Polym. Sci., Polym. Phys. Ed., 1983, 21:
2049.
[18] Panayiotou C, Vera J H. Polym. J., 1984, 16: 89.
[19] Marsh K N, Ott J B, Richards A E. J. Chem. Thermodyn., 1980, 12: 897.
[20] Gomez-Ibanez J D, Shieh J J C, Thorsteinson E M. J. Phys.Chem., 1966, 70: 1998.
[21] Ott J B, Marsh K N. Stokes R H. J. Chem. Thermodyn., 1981, 13: 37.
[22] Jain D V S, Lark B S. J. Chem. Thermodyn., 1973, 5: 455.
[23] Perez P, Valero J, Gracia M et al. J. Chem. Thermodyn., 1989, 21: 259.
[24] Gomez-Ibanez J D, Shieh J J C. J. Phys. Chem., 1965, 69: 1660.
[25] Shen W, An X Q, McElroy P J et al. J. Chem. Thermodyn., 1990, 22: 905.
[26] Vogel G L, Hamzavi-Abedi M A, Martire D E. J. Chem. Thermodyn., 1983, 15: 739.
[27] Gomez-Ibanez J D, Wang F T. J. Chem. Thermodyn., 1971, 3: 811.
[28] Nagata I, Ohta T, Nakagawa S. J. Chem. Eng. Japan, 1976, 9: 276.
[29] Li I P C, Wong Y, Chang S et al. J. Chem. Eng. Data, 1972, 17: 4923
[30] Apelblat A, Tamir A, Wagner M. Fluid Phase Equilibria,1980, 4: 229.
[31] Maciel M R W, Francesconi A Z. Fluid Phase Equilibria,1989, 50: 201.
[32] Harris K R, Dunlop P J. J. Chem. Thermodyn., 1970, 2: 805.
[33] Smith V C, Robinson R L. J. Chem. Eng. Data, 1970, 15: 391.
[34] Chaudhari S K, Katti S S. Fluid Phase Equilibria, 1989, 50: 329.
[35] Lark B S, Banipal T S, Singh S et al. J. Chem. Eng. Data, 1984, 29: 277.
[36] Chaudhari S K, Katti S S. Fluid Phase Equilibria, 1990, 57: 297.
[37] Kurihara K, Minoura T, Takeda K et al. J. Chem. Eng. Data, 1995, 40: 679.
[38] Jain D V S, Gupta V K, Lark B S. J. Chem. Thermodyn., 1973, 5: 451.