http://www.chemistrymag.org/cji/2001/032008pc.htm |
Feb. 1, 2001 Vol.3 No.2 P.8 Copyright |
----The p-p coordination effect on the stretching vibration frequency of the hydroxyl
Li Runqing, Fan Guoliang, Liu Cuihua, Li Yuhe
(School of Material Science and Engineering, Tianjin University, Tianjin, 300072)
Keyword Hydroxyl, Infrared Spectra, p-p p Coordination 有机结构波谱中二级结构效应机理的新探索(III)
----羟基伸缩振动频率中的p→p配位效应
李润卿
范国梁 刘翠华 李玉荷(天津大学材料科学与工程学院,300072,天津)
2000-07-12收稿
摘要
H2O和ROH中的O原子轨道为不等性sp2杂化的,并且其2p孤对电子和H原子的2p空轨道之间存在着一定程度的p→pp 配价键。这个配价键在O-H键伸缩振动频率随取代基电子效应的变化中起着主导作用。关键词 羟基,红外光谱,p→pp 配位
1.
问题的提出关于H2O和NH3的结构,目前比较一致的看法是:O和N原子轨道均为不等性的sp3杂化的四面体结构,O-H键长为95.8pm,N-H键长为100.8pm,键角∠HOH为104.5°,键角∠HNH为107.3°[1,2]。据此人们推论,烷基取代H2O或NH3中的H所得的醇或胺的O原子或N原子也是sp3杂化的[2]。酚、羧酸、芳香胺和酰胺的O或N原子为sp2杂化已是不争的认识。这就是说,在价电子的立体结构上,R-OH 和 R-NH2(含RR'NH)、Ar-OH和Ar-NH2 (含ArNHR和ArNHAr′)、RCOOH和RCONH2(含RCONHR′)(R为烷基,Ar为芳基),其O原子和N原子的价电子立体结构有着极大的相似性。若果真如此,那么O-H键和N-H键的伸缩振动频率随与它们相连的取代基的电子效应的变化规律应该是相似的。然而事实并非如此。
如表-1所示,-OH基伸缩振动波数随与之相连基团吸电子性能的增强,按醇、酚、羧酸的次序依次降低,而-NH2基伸缩振动波数则按饱和脂肪胺、芳香胺、酰胺的顺序逐次升高。 Table 1 The relations between X and n(OH) of X-OH and n(NH2) of X-NH2
表1 X-OH的n(OH)和X-NH2 的n(NH2 )与X的关系[3]
| X | -CH3 | CH3CO- | C2 H5CO- | Dn(a) | |||
| n(OH)/cm-1 | 3645 | 3638(b) | 3628 | 3618 | 3538 |
-100 | |
| n(NH2,as )/cm-1 | 3389 | 3381 | 3477(c) | 3475 | 3538 (d) |
157 | |
| n(NH2,s )/cm-1 | 3341 | 3316 | 3394(c) | 3390 | 3420(d) | 104 |
n(OH,酚) = -18.56s+3610.3 R=0.9444
n(NH2, as)= 40.74s+3477 R=0.9083
n(NH2, s) = 27.36s+3394 R=0.9137 Table 2 The relations between X and n(OH) of X
表2 X
| X | s[6] | n(OH,酸)/cm-1[7] | n(OH,酚)/cm-1[8] | n(NH2) | /cm-1[3] |
| p-NO2 | 0.78 | 3528.8 | 3593 | 3509 | 3416 |
| m-NO2 | 0.71 | 3529.6 | 3600 | ||
| p-C≡N | 0.66 | 3594 | 3505 | 3412 | |
| m-C≡N | 0.56 | 3495 | 3406 | ||
| p-COCH3 | 0.50 | 3502 | 3410 | ||
| m-COCH3 | 0.38 | 3486 | 3400 | ||
| m-Br | 0.39 | 3534.3 | |||
| m-Cl | 0.37 | 3534.5 | 3607 | 3490 | 3402 |
| m-I | 0.35 | 3533.8 | |||
| m-F | 0.34 | 3535.7 | |||
| p-Cl | 0.23 | 3535.5 | 3610 | 3482 | 3398 |
| p-Br | 0.23 | 3534.2 | |||
| p-I | 0.18 | 3533.4 | |||
| m-OCH3 | 0.12 | 3538.4 | 3485 | 3398 | |
| m-OH | 0.12 | 3497 | 3407 | ||
| H | 0.00 | 3539.2 | 3610* | ||
| p-CH3 | -0.17 | 3540.9 | 3612 | 3470 | 3390 |
| p-OCH3 | -0.27 | 3542.1 | 3614 | 3460 | 3382 |
| p-OH | -0.37 | 3617 |
Table3 The Influence of induced effect on
n(OH)表3 诱导效应对n(OH)的影响[3]
| Compounds(化合物) | CH3CH2CO2 H | ClCH2 CH2CO2 H | CH3CHClCO2 H | F3CCO2 H |
n (OH)/cm-1 |
3538 |
3534 |
3530 |
3504 |
再者,为什么对于分子间的作用-OH和-NH2却具有相同的变化方向?例如,氢键使-OH和-NH2的伸缩振动频率都发生大幅度的红移。 2.讨论
既然-OH基和-NH2基对取代基的电子效应的敏感程度及变化方向(波数升高或降低)不同,那就可以断定,O-H键和N-H键的结构一定有较大的差异。以H2O和NH3为例来讨论这个问题。NH3的键角∠HNH为107.3°,据此确认N原子轨道为不等性sp3杂化是毫无异议的,因为N原子只有一个杂化轨道被一对未成键电子占据。H2O的键角∠HOH为104.5°,如果仅仅据此来判定O原子轨道亦为不等性sp3杂化,则未必合理。这是因为O原子有两个杂化轨道被未成键电子对占据,如果这两个轨道相同,则O原子为不等性sp3杂化,如果这两个轨道不同,则为不等性sp2杂化。如表-4所示,1,1-二氟乙烯的键角∠FCF为109.2°,2-甲基丙烯的相应键角为109°。它们与sp3杂化的正四面体结构的键角(109.5°)极为接近,而且稍稍小一点,而与sp2杂化的正三角形结构的键角(120°)差别颇大。甲酸分子中的-OH基O原子与C=O基之间存在着p-p共轭,这已经是有机化学的常识。既然存在p-p共轭,那么 OH基O原子必须保留一个未杂化的p轨道。因此,羧酸分子中-OH基O原子应为sp2杂化。然而相关的键角为106.3°,不是接近120°,而是接近109.5°,而且还小一点。综上分析,可得出这样的结论:仅仅根据键角的大小来判断相关原子是sp2还是sp3杂化是不可靠的,也是不合理的。
Table 4 Several bond angles relate to hetero-sp2
表4 几种与sp2杂化有关的键角
| Compounds (化合物) |
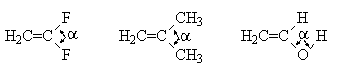 |
a |
109.2°[1] 109°[1] 106.3°[2] |
(HO)3P=O的P=O键并不是普通的双键。P原子轨道为sp3杂化的,其中一个杂化轨道上占有一对电子,它与O原子的一个空的2p轨道(假定是2px)形成一个σ配位键;O原子的2py和2pz轨道上各有一对电子,分别与P原子的3dxy和3dxz空轨道形成两个p→dp配位键[1]。(HO)2S=O的S=O键亦有与P=O键类似的结构[1]。
P原子的3d轨道能量为-2.25eV[1];S原子的3d轨道能量为-1.94eV[1]。它们与O原子的2p轨道能量(-13.6eV[1])差分别为11.35eV和11.66eV。H原子的2p轨道能量为-3.40eV,与O原子的2p轨道相差10.20eV,比P和O、S和O的相应值小。因此有理由认为,O-H键上存在着O原子的2p孤对电子与H原子2p空轨道间的p→pp配位键;在H2O中,每一个O-H键是由一个s共价键和“半个” p 配键组成的;在ROH中,其O-H键则是由一个s共价键和一个p→pp配位键组成的;而且从能量近似原则看,O-H键中的p→pp配位键要比P=O、S=O的p→dp配位键强一些。
另外,O-H键的键长(H2O和CH3OH为96pm)[1]比P=O键的键长(PO43-的这一键长为156pm)[1]短得多。这就为O原子和H原子的两个2p轨道有效的“肩并肩”地交盖形成p配位键提供了有利的条件;同时O-H键如此之短,这本身或许亦表明它不仅仅由一个s共价键组成,还存在着一定程度的p→pp配位键。
假定O-H键的O原子轨道为sp2杂化,且在该键上存在 着p→pp配位键,那么将会产生什么效果呢?
对于含有孤对电子的原子,如O、N等原子来说,有两个相反的因素影响着原子轨道的结构:(1)为了使原子轨道成键能力最大,原子轨道必须杂化;(2)为了避免s电子的激发,原子轨道最好不杂化。O原子的2s轨道能量为-28.5eV,2p轨道能量为-13.6eV,两者相差14.9eV。如果采取不等性sp2杂化,则占有一对孤对电子的杂化轨道保持有较多的s成分,这既可以尽可能地避免s电子的激发,又可以使两个各占有一个电子的杂化轨道的成键能力得以提高,同时未参与杂化的一对p电子还可以和H原子的2p空轨道形成一定程度的p配位键,使该对电子得以稳定。如果采取sp3杂化,那将失去p→pp配位产生的稳定化作用。因此有理由认为,从整体价电子的稳定性考虑,在H2O和ROH中,O原子轨道采取不等性sp2杂化比采取不等性sp3杂化更为有利。
我们已经另文讨论了化学键伸缩振动频率与取代基电子效应间的关系(《光谱学与光谱分析》,2000年第5期):对于X-A-B(A-B表示以共价键将原子A和B结合在一起,它们之间可以是单键,也可以是多重键;A和B可以相同;X为取代基)来说,吸电子能力强(电负性大或s 值大)的X使A-B键增强,因而伸缩振动频率升高;供电子能力强的X使A-B键的强度减弱,因而伸缩振动频率降低。对于X-A→B(A→B表示A和B以配位键结合在一起,A为提供电子对的原子,B为提供空轨道的原子;X为取代基)来说,吸电子能力强的X使A原子提供电子对的能力减弱,因而A→B配位键减弱,其伸缩振动频率降低;供电子能力强的X使A原子提供电子对的能力增加,因而A→B配位键强度增加,其伸缩振动频率升高。对于A→B-X来说,A→B键的伸缩振动频率和X的电子效应间的关系则正相反。
对于N-H键来说,因为它是一个正常的s键,故其伸缩振动频率理应随与之相连的取代基吸电子能力的增加而升高,这表现在以下几个方面:
(1)取代基电负性的影响 烷基胺的N原子轨道为sp3杂化,当与N原子相连的烷基对N H键的位阻作用差别不大时,N-H键的伸缩振动频率就仅由它们的电负性大小决定,电负性越大,振动频率越高。这就是(F3C)2N-H的N-H伸缩振动频率比(CH3)2N-H的高的主要原因。
(2)N原子轨道杂化状态的影响 芳香胺的N原子轨道为sp2杂化。由于未参与杂化的2p孤对电子和芳香环之间的p-p 共轭使p电子对向芳环转移,N原子周围的电子云密度降低,对N-H键的σ电子吸引力增强,因而N-H键强度增加,伸缩振动频率升高。这就是芳香胺的N-H键伸缩振动频率比烷基胺的高的主要原因。与芳香胺相比,酰基的吸电子能力强得多,故酰胺的N-H键伸缩振动频率又比芳香胺的高。
需要说明的是,在芳香胺、酰胺中,N原子的未参与杂化的2p电子对似乎亦应与H原子的2p空轨道形成p→pp 配位键,但因为N原子的2p轨道能量较低,为-14.5eV(O原子的2p轨道为-13.6eV),与H原子的2p轨道能量差较大,故在芳香胺中的N-H键上的p→pp 配位键的贡献很小(与O-H比),似乎可以忽略不计。
(3)s值的影响 芳香环上的取代基的电子效应是其电负性和共振效应的综合结果,其吸电子能力的大小可用其Hammett常数s表征。H原子的s为零;吸电子能力比H原子强的基团的s为正值;吸电子能力比H原子弱的基团的s为负值。表2所列苯胺的-NH2 基伸缩振动波数表明:吸电子取代基使-NH2的伸缩振动波数升高;供电子取代基使其伸缩振动波数降低。
对于O-H键而言,由于存在着p→pp 配位,故可写成O
R-OH 和Ar-OH中的O原子轨道都是sp2杂化的,这已在上文进行了详细的论述。Ar-OH中的p-p 共轭虽然会使O-H键中的σ 键增强,但同时也会使p→pp 配位键减弱。由于p 电子云的流动性比s电子云大得多,故p→pp 配位键减弱导致的O-H键的减弱,远比s 键增强的贡献大,总的结果是O
另一个令人振奋的信息是,上述四个Hammett方程中的r值。|r|值说明了振动频率对取代基电子效应的敏感程度。苯胺的-NH2的|r|值(反对称伸缩振动为40.74 ,对称振动为27.36),比酚的(|r|=18.56)和取代苯甲酸的(|r|=12.49)大得多,这是为什么呢?
对N-H键的强度来说取代基只有一种作用:或使其增强,或使其减弱。对O-H键而言,取代基同时发生两种作用:或使s 键加强和p→pp 配位键减弱,总的结果是使O-H键减弱;或使s键减弱和p→pp 配位键加强,总的结果是使O-H键加强。 红外光谱所表现的是两种作用的代数和,故O-H键的|r|值比N-H的小。类似的规律在表-1所列数据中亦有体现。同理,因为苯甲酰基的吸电子能力比苯基强,故与酚相比,苯甲酸的O-H键中的p→pp 配位键的贡献要小一些,因而取代苯甲酸的|r|值比酚的小一些。
对于取代基的电子效应,O-H键和N-H键有着迥然不同的表现,已如上述,但对于分子间的作用,却有着相同的表现。例如,氢键使它们的伸缩振动吸收带均发生大幅度的红移。这又是什么原因呢?O-H键和N-H键均为强极性键,O或N原子为负极,H原子为正极。当它们与其他强极性基团形成氢键时,虽说O或N原子和外来强极性基团的正极缔合的同时,它们的H原子亦和外来基团的负极缔合,但对于O-H或N-H键伸缩振动频率的影响,主要来自H原子和外来基团负极的缔合作用,这已得到证明[9]。与O-H或N-H键的H原子缔合的负极带负电,负电对O
3.
结论综上分析,可以得出如下结论:H2O、ROH、ArOH、R-COOH和Ar-COOH中的O原子轨道均为不等性sp2杂化的,而且在O-H键中存在着一定程度的p→pp配位。虽然这个p配位键对O-H键强度的贡献是次要的,但由于p电子云的流动性远大于s电子云,故在O-H键伸缩振动频率随取代基电子效应的变化中,p配位键却起着主导作用。 REFERENCES
[1] Xu Guangxian, Wang Xiangyun. Matter Structure. 2nd ed. Beijing: Higher Education publishing house, 1987.
[2] Xu Shouchang. Organic Chemistry. Beijing: People's Education Publishing House, 1982.
[3] Xie Jingxi. The Application of Infrared Spectroscopy in Organic Chemistry and Medical Chemical. Beijing: Science Publishing House, 1987.
[4] Lu Yongquan, Deng Zhenhua. Applied analysis for Infrared Spectra. Beijing: Electron Industry Publishing House, 1989: 8.
[5] Bellamy L J (Huang Weiyuan, Nie Chongshi translated). The Infrared Spectra of Complex Molecules. Beijing: Science Publishing House, 1975: 250.
[6] March J. Advanced Organic Chemistry. New York: McGraw-Hill, 1970: 253.
[7] Li Runqing, Li Yuhe, Chen Huiming. Structure Chemistry (Jiegou Huaxue), 1989, 8 (4): 294.
[8] Zhao Yaoxing, Sun Xiangyu. The Analysis of Spectra and The Identify of Organic Structure. Hefei: Science and Technology University of China Publishing House. 1992: 134.
[9] Li Runqing, Zhang Jie, Li Yuhe. The Study on Mechanism of Solvent Effect in Infrared Spectra (III). Journal of Tianjin University. 1993, (5): 94.