http://www.chemistrymag.org/cji/2001/039045re.htm |
Sep. 1,
2001 Vol.3 No.9 P.45 Copyright |
Recent advances in organometallic amphiphiles
Wang Chuanzhong, Huang
Jianbin, Tang Shihua
(Institute of Physical
Chemistry, Peking University, Beijing 100871, China)
Received Mar. 2, 2001; Supported by the Natural
Science Foundation of China (20073002,29992594) and Research Fund for the Doctoral Program
of Higher Education
Abstract Organometallic
amphiphiles are new class of surfactants. Various novel chemical and physical properties
of their aggregates, such as selective binding to metal ions, active control of
interfacial tension, catalytic behavior and charge transfer, have been widely investigated
in recent years. These interesting properties are reviewed in this paper.
Keywords
Organometallic amphiphile, Surfactant, Aggregation
The research of organometallic amphiphiles is interesting and important, sinceˇˇ it combines several areas of modern science such as organometallic
chemistry, coordination chemistry and surface science. Organized molecular assemblies such
as micelles, vesicles and monolayers formed from organometallic amphiphiles, have
attracted many chemists' attention for constructing supramolecular catalytic system,
growing size-quantized semi-conductors and other functions. It was thought that the
research around organized assemblies from organometallic amphiphiles is a promising work
due to their unique properties, including selective binding to metal ions, active control
of interfacial tension, catalytic behavior, charge transfer, etc. In this article an
overview of them is presented to broaden the knowledge of us.
2. SELECTIVE
BINDING TO METAL IONS
Recently, organized assemblies containing metal ions have attracted much attention
for selective binding to metal ions and developing ion sensors from both fundamental and
practical viewpoints. Most of interests are devoted to interaction between monolayers and
metal ions in the subphase. Apart from the widely used fatty acids[1], some
amphiphiles functionalized by crown ether[2], imidazole[3] and
iminodiacetate[4] have been designed as monolayer-forming reagents and their
interactions with metal ions in the subphases have been studied.
ˇˇ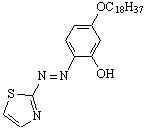
Figure 1. The Structure of TARC18
Table 1. Metal Contents
in the Scraped Films from the Monolayers on Aqueous of Metal Ions (ppm)a)
ˇˇ |
Fe(II) |
Co(II) |
Ni(II) |
Cu(II) |
Zn(II) |
Cd(II) |
subphase I |
0.36 |
0.80 |
0.66 |
5.82 |
0.85 |
0.63 |
subphase II |
0.24 |
0.14 |
0.25 |
4.85 |
0.28 |
0.28 |
a)
Subphase I: mixture of each 0.5 mM Fe(II), Co(II), Ni(II), Cu(II), Zn(II), and Cd(II).Subphase II: mixture of each 0.1mM Fe(II), Co(II), Ni(II), Cu(II), Zn(II), and Cd(II).
Another surface-active molecule, Kemp's triacid, has been identified as a complexing agent for heavy metal ion, such as lead and copper[6] (Figure 2). This unique molecule may be utilized for removal of toxic metal and metal-complexation properties.
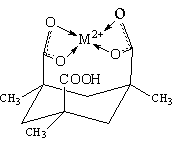
3. ACTIVE
CONTROL OF INTERFACIAL PROPERTIES
Switchable control of bulky solution properties of surfactants by physical or chemical method (such as chemical redox, electrochemical redox, light, etc) has attracted considerable interest from various viewpoints. The experimental experience in the study of redox-reversible system leads chemists to suspect that similar principles could be applied to aggregation behavior. In particular, a compound with two non-polar ends may be converted into an amphiphile by reaction of one end, if chemically switchable, with either an oxidizing or reducing agent. The resulting altered structure, if amphiphilic, might then aggregate under appropriate conditions. Such a possibility was explored by Graetzel and his co-workers who prepared an amphiphilic cyclam derivative rendered "swithable" by incorporation of a copper ion[7]. These novel organaometallic amphiphiles formed micellar aggregates.
S. Munoz and G. W. Gokel synthesized a nickel(II) phenanthroline complex 1 (Figure 3)[8] to form vesicles in aqueous solution. The vesicles were characterized by dynamic light-scatting technique. When aqueous solution of Ni complex in Tris ·HCl buffer was treated with an aqueous solution of Na2S2O4, the vesicles collapsed completely. It is because that the reduction of transition metal complexes of phenanthroline occurs at the ligand rather than at the metal center. Two electrons reduction gives an intramolecular ion pair of inherently low charge density. Reduction of the hydrophilic headgroups diminishes or eliminates monomer amphiphilicity and ultimately leads to deaggregation of the assemblies.
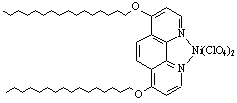
Figure 3. The complex of Ni2+ and twin-tailed phenanthroline derivative
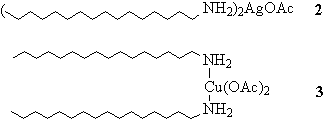
Figure 4. The metal-hexadecylamine complexes
In the meantime, the principles for in situ and reversible control of the interfacial properties of surfactant-based systems were established in order to make possible active control of phenomena such as the stability of thin films of liquid, the spreading or wetting of fluids on solid surfaces, and the transport of solutes across interfaces.
N. L. Abbott et al reported a method for active control of the surface properties of aqueous solutions by using electrochemical techniques in combination with redox active ferrocenyl surfactants Fc(CH2)nN+(CH3)3Br- (n=8 [I+],11,15)[9] (Scheme I).
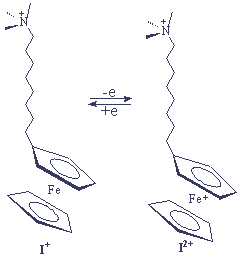
ˇˇˇˇˇˇˇˇˇˇ Scheme I
Electrochemical oxidation of ferrocene to ferrocenium (or reduction of ferrocenium to ferrocene) caused reversible changes in surface tension as large as 23mN/m.
A dimer, ferrocenyl surfactant (DI2+) (Scheme II) with more unique properties have also been synthesized.
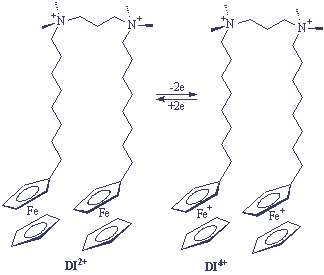
ˇˇˇˇˇˇˇˇˇˇˇˇ Scheme II
The surface activity and micellization of DI2+ were compared and constructed to those of the monomer I+ in aqueous solution. The experimental results showed that oxidation of DI2+ to DI4+ made possible changes in surface tension of 21mN/m over a range of concentrations that exceeded an order of magnitude (0.02-0.4mM). Control of the oxidation state of I+, in contrast, permits changes in surface tension of about 20mN/m at one concentration (2.0mM) only (Figure 5).
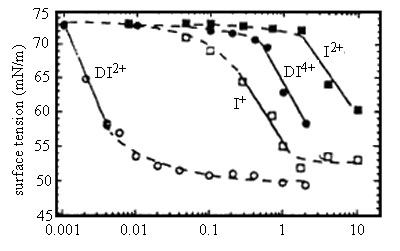
Figure 5. Equilibrium surface tensions of aqueous solutions of dimeric and monomeric ferrocenyl surfactants in oxidized and reduced states: DI2+; DI4+; I+; I2+. All measurements were performed in aqueous solutions of 0.1M Li2SO4 at 25ˇăC and pH 2.0
Dimeric redox-active
surfactants in combination with electrochemical method do, therefore, permit active
control of interfacial properties of aqueous solution over a wider range of surfactant
concentrations than is possible with the corresponding monomeric redox-active surfactant.
Gradients in surface tension, established
by electrochemical formation of surface active species in spatially localized (smaller
than millimeter) regions, have been used to direct the motion of fluid across the surfaces
of solution.
4. CATALYTIC BEHAVIOR
Micelles of amphiphiles often catalyze chemical
reactions, which have been attracting chemists' attentions.[10] Recently there
is continuous interest in the hydrolysis of phosphate ester, because that 1) highly toxic
derivatives belong to this class of compounds, and 2) nucleic acid bases are hold together
by phosphate bonds. Powerful catalysts of the cleavage of these esters may be exploited as
useful detoxification reagents or as antibiotics as well.
Organometallic amphiphiles proved
particularly effective in the cleavage of lipophilic triester in aqueous solutions where
they form micellar aggregates (metallomicelles). One of the most effective metallomicellar
systems, introduced by Menger in 1987[11], is based on the Cu(II) complex of
N-n tetradecyl-N,N'-N'-Trimethylethylenediamine(TMEDC14) 4 (Figure 6).
It is reported that TMEDC14 possesses a remarkable
catalytic activity in hydrolysis of phosphotriester. The 4-promoted hydrolysis is more
than 200 times faster than the hydrolysis of 5 catalyzed by an equivalent concentration of
a nonmicellar homologue, CuII-tetramethylethylenediamine complex 6. They also
showed that nerve gases, such as soman 7, were very efficiently detoxified with 4 at pH
7.0 (detected by the release of F-). The possible reasons for the rate
accelerations were the enhanced electrophilicity of the micellized copper(II) ion, the
acidity of CuII-bound water, or the intramolecular type of reaction due to the
micellar formation. The CuII-OH- species 4b were
postulated to be an active catalytic species. On the basis of the rate-pH profile for pH
6.0-8.3, the pKa value for the acidity of CuII-OH2(4a)![]() CuII-OH-(4b)
+ H+ was estimated to be less than 6.
CuII-OH-(4b)
+ H+ was estimated to be less than 6.
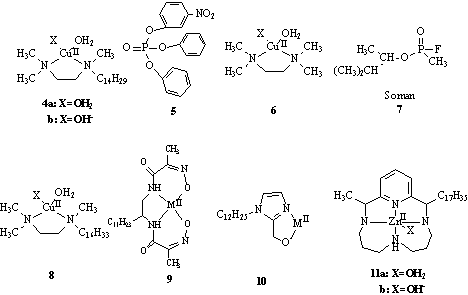
Figure 6. The structure of catalysts and substrates
R.A. Moss et al[12] have
performed a deeper research work on this area, and prepared an organometallic amphiphile
TMEDC16.Cu(II) 8. Compared with monomeric metallocatalysts,
E. Kimura[16] and his coworkers synthesized a cyclen
derivative attached to a long alkyl chain(C16H33), as shown in
Figure 7. Anticipated supramolecular assemblies between the zinc(II) complex and
lipophilic substrates that coexist in micelles could make hydrolysis of lipophilic ester
more effective. The experiment revealed that the active species was estimated to be ZnL-OH-
rather than ZnL-OH2, and the hydrolysis of TNP is 290 times more efficient than
with ZnL'-OH-.
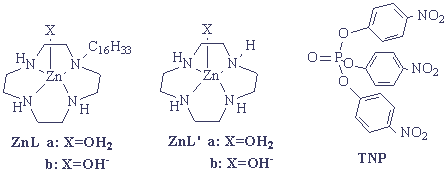
Figure 7.
The structure of ZnL, ZnL' and TNP
Other catalytic systems
were also investigated. For example, H.Chen et al [17] studied olefin
hydroformylation by water-soluable rhodium complexes with TPPTS
(tri(m-sulfonphenyl)phosphine) as ligand.
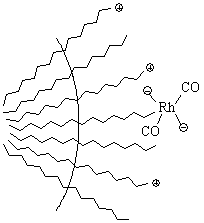
Figure 8. Rhodium complexes in micelle
Their study demonstrated that 1-dodecene
hydroformation in biphase catalytic system occurred in the interface of aqueous/organic
phase. The formation of micelle was not only favorable for the reaction acceleration, but
also favorable for the increase of linear aldehyde ratio in products. The key factor of
the enhancement of reaction rate was the richness of rhodium electricity attraction
between active rhodium anion species and cation end of surfactant.
5. ELECTRON
TRANSFER REACTION
There is much current interest in the design and study of vectorial photoinduced electron systems using micelles and vesicle membrances in aqueous media. In these systems, the photoactive centers, namely synthetic porphyrin derivatives, play a crucial role,[18] which has been widely reported.
T. Komatsu[19] reported a bolaamphiphilic meso-tetrareesorcinolporphyrins with eight octadecyl-w-phosphocholine side chains (octopus porphyrin, Figure 9(1)), which formed either monolayers or open fiber structruct. The fibers fluoresced strongly, and laser flash photolysis lead to electron transfer from the porphyrin to hydrophobic phenyl-p-benzoquinone as well as to hydrophilic 1,2-naphtoquinone-4-sulfonate and dimethyl viologen acceptors.
Photoinduced electron transfer between porphyrins has also been investigated. T. Yanagimoto[20] found significant photoinduced electron transfer reactions between lipidporphyrinato-zinc(II) (12) and iron(III) (13) complexes (Figure 9(2)) in their molecular assemblies. The self-assembled micelles of 12 and 13 showed negligible fluroscence of the photodeactive complex between the two chromophores in the ground state. On the other hand, 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (abbreviated as DMPC) vesicles embedding the two chromophores at moderate ratios represented photoinduced electron transfer. Because of the low mobility and high dispersibility of the lipidporphyrins anchored into the gel state of DMPC membrance, the Stern-Volmer plots for the fluorescence lifetime showed a straight line up to high concentrations of the quencher; the quenching rate constant was estimated to be 1.2ˇÁ1011 M-1s-1. The electron transfer presumely occurs by a tunneling mechanism when the nearest neighboring 13
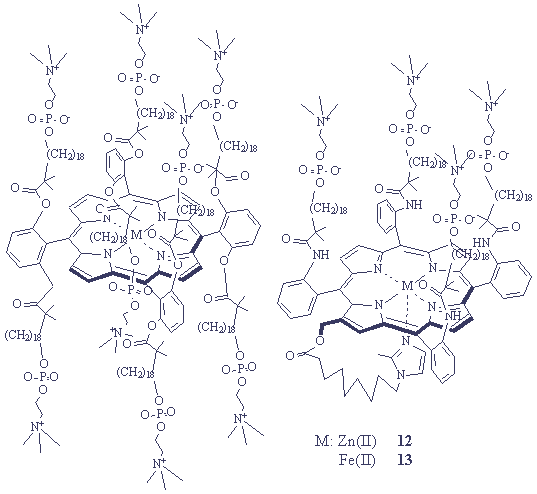
ˇˇˇˇˇˇˇˇ ˇˇ (1) ˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇ (2) ˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇˇ
Figure 9. The structure of (1) octopus and (2) lipidporphyrin
6. CONCLUSIONS
Some properties of these organometallic amphiphiles, such as selective binding to
metal ions, active control of interfacial tension and catalytic behavior, are present in
this article. However, the unique properties of this kind of compounds are far more than
those noted, if considered the introduced functional groups. Since the beginning of 90s,
organometallic amphiphiles have been becoming more and more accessible. Functionality of
increasing complexity has gradually be introduced, leading to their organized assemblies
displaying novel chemical and physical properties in relation with coordination chemistry,
electrochemistry and molecular meterials. Undoubtly
REFERENCES
[1] Furlong D N, Urquhart R, Grieser F et al. J. Chem. Soc., Faraday Trans., 1993, 89 (12): 2031.
[2] Fainerman V B, Miller R, Aksenenko E V et al. Langmuir, 2000, 16 (9): 4196
[3] Van Esch J H, Nolte R J M, Ringsdorf H et al. Langmuir, 1994, 10 (6): 1955
[4] Shnek D R, Pack D W, Sasaki D Y et al. Langmuir, 1994, 10 (7): 2384
[5] Liu M, Kira A, Nakahara H. Langmuir, 1997, 13: 779
[6] Sengupta T, Yates M, Papadopoulos K D et al. Colloid and Surfaces A, 1999, 148 (3): 259
[7] Humphry-Baker R, Moroi M, Gratzel M et al. J. Am. Chem. Soc., 1980, 102 (11): 3689
[8] Munoz S, Gokel G W. Inorg. Chim. Acta, 1996, 250: 59
[9] Gallardo B S, Hwa M J, Abbott N L. Langmuir, 1995, 11: 4209
[10] Romsted L S, Bunton C A, Yao J H. Current opinion in colloid & interface science, 1997, 2 (6): 622
[11] Salmon A, Dodd S W, Eagerton F D et al. J. Am. Chem. Soc., 1987, 109 (9): 2600
[12] Scrimin P, Ghirlando G, Tecilla P et al. Langmuir, 1996, 12 (26): 6235
[13] Gutsche C D, Mei G C. J. Am. Chem. Soc. 1985, 107 (26): 7964.
[14] Tagaki W, Ogino K, Tanaka O. Bull. Chem. Soc. Jpn., 1991, 64 (1): 74
[15] Gellman S H, Petter R, Breslow R. J. Am. Chem. Soc., 1986, 108 (9): 2388
[16] Kimura E, Hashimoto H, Koike T. J. Am. Chem. Soc., 1996, 118 (45): 10963
[17] Chen H, Li Y, Chen J et al J. Mol. Cata A, 1999, 149 (1): 1.
[18] Kaneko M, Tsunchida E, Imai Y et al. J. Chem. Soc. Faraday Trans., 1991, 87 (1): 83.
[19] Komatsu T, Yamada K, Tsuchida E et al. Langmuir, 1996, 12 (26): 6242
[20] Yanagimoto T, Komatsu T, Tsuchida E. Inorg. Chim. Acta., 2000, 305 (1): 26.
ˇˇ