http://www.chemistrymag.org/cji/2003/051001pc.htm |
Jan. 1, 2003 Vol.5 No.1 P.1 Copyright |
Xia Bingle,
Peng Dungeng, Liu Qingliang
(Department of Chemistry, University of Science and Technology of China,
Hefei 230026)
Abstract On the basis of reading a large
number of articles, we reviewed the development of the research in mechanism and
application of several peroxidases. We made a compare the commonness and individuality of
the peroxidases in the mechanism and the application. The insufficiency in the researches
was pointed and a prospect of the researches, which we thought were necessary in these
fields in the following years, was made in this review.
Keywords peroxidase, reaction mechanism, application, development
(�й���ѧ������ѧ��ѧϵ,�Ϸ�,230026)
2002��9��16���ո壻�����̲�ר��ֿ��л����������⡣
ժҪ
�ڱ��������ǽ�������Դ�Ĺ�������ø�Ŀ��ܵķ�Ӧ����������������ǵĽṹ�Լ�Ӧ�������������ڽ��ܷ�Ӧ������Ӧ�õ�ʱ��չʾ�����ǵĸ��ԣ����ܽ������ǵĹ��ԣ�ָ����Ŀǰ�о������IJ��㲢�Խ���������ø����Щ������о�����չ����
�ؼ��� ��������ø����Ӧ������Ӧ�ã���չ
��������ø
(Peroxidase,EC1.11)�ǹ㷺�������������е�һ��ø,�ֺ���Ѫ���أ���һ�ֽ�ϵ��ס������Դ�H2O2������л��������������л������������������Ӧ[1-5]�����Ƕ�����ø�ķ��봿������Ӧ�������ṹ��Ӧ�ö�������Ӧ���о���Ŀǰ����������ø��ø�Ĺ��������߲ⶨ������ҩ����ϼ�����Ӧ�ý�Ϊ�㷺������Ӧ�������ﴫ������ҩ��ĸĽ�����ѧ�Լ������������㻯����Ľ��⣬������⣬�ڲ��Ͽ�ѧ����Ҳ��Ӧ�ã��������������ȩ���ܼ�����֬Ϳ�ϣ��������������ɷֵ�øѧ�ⶨ������������������ϡ�������ֲ���������ø��ͬ��ø�������ձ�����ʺ������������ı�ɫ��������ʹ�ù�������ø��Ϊһ�ֱ�����ø�����ڻ���ѧ������ѧ�Ͳ���ѧ�о��з�������Ҫ���á���������ø��������Ӧ��������չ���ײ���Ķ������ӡ���������������������Լ�ϸ�����������ĵ��ڡ�
�����Ϲ�������ø�Ľṹ�������ǵķ�Ӧ������������ʵ�������е�Ӧ�������棬��������������Щ�����ϵĽ�չ�� 1. ����
��������ø�Ĵ�����һֱ�������о���һ���ȵ㣬���ܹ�ע��ԭ���ǻ���������ľ�ʻ���ϸ���ֻ���ϸ�����źŴ��ݡ�IAA�³´�л�������Լ��������п��ܰ��ݼ�����Ҫ�����á������Ϲ�������ø�ĵĽṹ�������Ӧ�������о���չ��һ���ܡ�
1.1 HRP�Ĵ�����
HRP�����ǽ������ú��о��Ĺ�������ø�������������������ø���������о���Ϊϵͳ����ϸ�����¾����ڷ�Ӧ��Ӧ���е����û�����һ���ܡ�HRP��X����ṹ����1997�걻����������ʾ����������ͬһ���壨�������壩�Ļ�����������ø�������������ø��һ����ͬ�㣬����Ѫ���صĿڴ�����pocket����Ϊ���˺�Զ�˵��������֣������۵��а��������������ӡ�
Anderson���� [1]�ö����ų�ЧӦ�о��� HRP���м仯���� ,��Ϊ HRPѭ����Ӧ������ṹ����ͼ1��ʾ .���������������������ø��ľ������ڷ�����Һϵͳ�н����˹��۷�Ӧ��ʵ����ʾ�ı��ڲ�������ṹ[2]��
���ڷ�Ӧ�м���Ĵ�����ӹ���ɣ�����ʵ���ֶζ�֤�������Ĵ���,���м��嵽���Ǻ��ֽṹ��ȴ��˵��硡�Dunford,Haesun���˶���Ϊ�����Ժ�������Һ�л����� I ���γɹ���������ʾ:
H2P��HP��P+H2O2��HRP-I
��ͬ���ǣ��������ٿز�����������,������Ϊ���γ�HRP-I֮ǰ����һ�������ֿ��ܽṹ�Ļ�����0�Ĵ��ڣ�������˹�������ø��ת�����裬��ϸ���ݿɲ�������[3]��
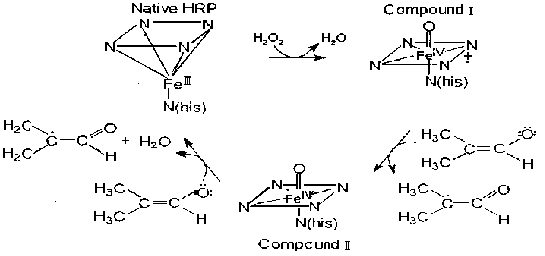
ͼ1 ������������ø�Ĵ�ѭ��
1.1.2 HRP
����缫�ϵ���������ԭ������������ø���ε缫�� H2O2���㰷�����ࡢ�����ǵ����ʵIJⶨ�����ź���Ҫ������ ,���Խ������õ��˽�Ϊ�㷺���о��ͷ�չ[4]��
HRP�ڵ缫�����ϱ������γ��м仯���� I �� II, H2O2ֱ�Ӵӵ缫�ϵõ����Ӷ�����ԭ (ֱ�ӵ���ת�� ),���ߴ�������ԭý�����ϵõ����Ӷ�����ԭ (��ӵ���ת�� ),�����ֵ���ת�Ʒ�ʽҲ����ͬʱ���� [5],���ַ�ʽ�õ��Ļ�ԭ���������������Ũ���йء����� ,����������Ũ�Ⱥܸ�ʱ ,HRP�����γ�һ�ֲ����õ���ʽ������ (����̬Ϊ +6).������ HRP���ε缫 ,���ڻ���缫����������һ�� HRP���ӹ��ɡ�HRP�缫�ڴ�ѭ�������з����ķ�Ӧ����:
HRP(Fe3��)+H2O2 ��Compound-I (1)
Compound-I +AH2 ��Compound-II +AH· (2)
Compound-II +AH2 �� HRP(Fe3+) +AH· +H2O (3)
���Ѵ˵缫���������Ʒ�� ,�����ֵ缫��λ����600m V(SCE)ʱ ,HRP������Ӧ���м仯����ӵ缫�ϵõ����Ӷ�����ԭ ,��ԭ������H2O2��Ũ�ȳɱ��� ,ͬʱҪ��֤��Һ�б�����H2O2���ڡ������������£�HRP��缫֮��ĵ�����ֱ��ת�Ƶ�[4]��
HRP��Ӧ�м��廯����I�ͻ�����II�������Ļ�ԭ��ʽ ,�Ǵӵ����ϵõ����ӡ�HRP����Ӧ�γɵ���������ȡ���ڵ��� (���ӹ��� )�����ʡ����㰷�༰����ȵ��ӹ��� ,���������γ����ɻ� AH·;�еĵ����������������II(hexacyanoferrate II)ʧȥһ�����ӻ������ʧȥ�������Ӻ� ,�����γ����ɻ������ӹ��屻�������γɵIJ��� (���ɻ� AH· ),���ٷ������·�Ӧ[5]:
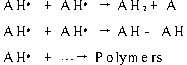
����Fe3+Ϊ HRP�ĸ����� ,AH2Ϊ���ӹ��� ,��Ϊ���㰷�������� ,AH·Ϊ��������������ɻ�����Ӧ �� HRP�� H2O2��ͷ��������л������������� ,ʧȥ�������������м仯����I (����̬Ϊ +5�� ),���м����ɺ�����(Fe4+=O)��һ��߲����������� ;�����ķ�Ӧ�� ,������ I �ӵ�һ�����ӹ���AH2�ϵõ�һ������ ,����ԭΪ�м仯����II (����̬Ϊ +4�� );�����һ����Ӧ�� ,�м仯����II�ӵڶ������ӹ��� AH2 ���ֵõ�һ������ ,���� HRP����ԭΪ��ʼ״̬��Annika Lindgren���˱����˹���������HRP��TAP���εĵ缫�ϵ�ֱ�ӻ�ԭ���̣����������������绹ԭ������HRP�Ĵ����ܸ���[4]��
1.2 ���鶯���������ø(Mammalian peroxidase)
��ֲ���������ø����Դ���ƣ����鶯���������øҲ�Ǵ�һ��Զ������ݱ�����ġ����ڼ�״�ټ��صĺϳɺ͵��������з�������Ҫ����[6]��
�ڲ��鶯���������ø���о��϶���м�״�ٹ�������ø��TPO��thyroid peroxidase�������������ø��MPO,myeloperoxidase��,�����ϸ����������ø(EPO,eosinophil peroxidase),���������ø��LPO,lactoperoxidase����
��������鶯���������ø(P)������ͨ�����·�Ӧ���������ӵõ��ε���͵⣺
P compound I +I�� ��POI�� (1)
POI��+H����P+HOI��2��
POI��+I����P+I2+OH- (3)
�Ұ�������������ʵ���ø�⻯��HOI��I2�������º���������Ȼ��ͨ����������ø-�ε����θ�������̣���ø����ĵ⻯������LPO�Ȳ��鶯���������ø�з�����
POI��+Tyr+H����ìmonoiodotyrosine(MIT)+LPO+H2O (4)
POI��+MIT+H����ìdiiodotyrosine(DIT)+LPO+H2O (5)
1.3�̲ݹ�������ø���������о���չ
ͬ����ά��ֲ��һ�����̲��������Ӵ�Ĺ�������ͬ��ø�����Ҷ���Դ�����е��ӷ�Ӧ��������Ϊ����ʹ��H2O2��O2 ��Ϊ���ӽ����尴�յ��������̲��뷴Ӧ�����̲ݹ�������ø�Ĵ�������δ�γɶ���. �й۵���Ϊ��HRP�Ĵ���Ӧ��,���ɻ����̶������������˹��ף�����TOP��������ȴ���ɻ�����I��������Ҫ�Ľ�ɫ[7��8]��
1996��Irina G.Gazarianͨ�����������ᣨIAA�����о�����÷�Ӧ�е�ø��ѭ�����̣�
E3++IAA�� E2++IAA��+ (1)
E2++O2�� compound III (2)
Compound III+IAA��E2++CO2+H2O+indole-aldehyde(������ȩ) (3)
��һ�ֿ��ܵ����û�����ͼ2��ʾ��
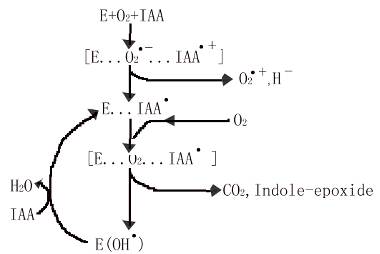
Fig.2 The mechanism of the reaction of O2 with IAA catalyzed by heme peroxidase
1998
��Gazarianͨ����������������HRP��TOP��IAA��O2�Ĵ���Ӧ����ѧ���о��������ʾ������Ѫ���ص�ֲ���������ø������û�������Ĺ�����������������ɴ���Ӧ����ȱ��H2O2���������ֲ���������ø��O2��IAA�ķ�Ӧ��һ������Ҫ��������Ӧ��IAA�ڹ����������ɻ����µĽ��ⷴӦ���1955��Ϳ�ʼ�������о���������������۸��ӵIJл���Ӧ�����䷴Ӧ����,�������ͳһ����Ϊ�ձ�Ĺ۵��ǣ�1������6��E��EI��EII Ϊԭø����Ӧ�ĸ����IAAOOH ��Դ��IAA��IAAOH�����Ӧ�Ĵ�
IAA+O2�� IAA.+�� ��1��
IAA.+ O2�� IAAO2. ��2��
IAAO2. +IAA +? �� IAAOOH+IAA. ��3��
E+ IAAOOH�� EI +IAAOH ��4��
EI +IAA�� EII +IAA. ��5��
EII+ IAA�� E +IAA. ��6��
Irina G.Gazarian�Ѿ�������[7]�в����ˣ����ȶ�״̬�£�TOP��IAAOH�����ܵ���������ø��Ӱ�죬��֪HRP����Ӧ���������ø�ء������ڶ�TOP/HRP��GANSIENT����ѧ�о��л�����I��û�й۲쵽.
�ڶ������HRP����Ļ����ǽ����ڻ�����III�IJ�������ϵģ�
E+ IAA�� E2+ +IAA.+ ��7��
E2++ O2�� EIII ��8��
EIII+ IAA�� E2++ IA +CO2+ H2O ��9��
EIII+ 2IAA�� EII +2IAA. ��10��
EII+ IAA�� E-IAA.+ H2O ��11��
E-IAA. +O2�� EII+In-CH2O +CO2 ��12��
����[7]��Ϊ�����������ø��IAA ��Ӧ�е�ʽ��7���Dz����ڵ�,��Ϊû���㹻��֤�ݱ������ֲл��γɵĿ����ԡ��������������Ҫ�IJл��Ĺ��̻��Dz������
Inna G. Gazaryan����Ϊ��Ҫ�IJл������ɣ��������������Ӵ���ʱ��IAA����ʱͬʱ�����л���������ø�������γɵ�[8]:
E+ O2+ IAA �� [IAA-E-O2] ��13��
[IAA-E-O2] �� E +IAA.+ +O2.- ��14��
1.4 LiP(Lignin peroxidase)
LiP
ľ���ع�������ø��LiP����343��������л���370��ˮ���ӣ�Ѫ���أ�4���Ǻ�������������ɡ�ľ���ع�������ø��ʾ�˵��͵Ĺ�������ø�Ŀռ��۵����������Ѫ������һ������������������ø��һ���ķ�ջ�����close environment�������б�����Trp171�������ڵڶ���������λ�㣬����ѱ����������Ĺ�������ø�ľ���Ѫ���ر�Ե��ͬ��Itakura��Henriksen�ֱ�Ծ����Ѫ���ر�Ե���������Ĺ�������ø��Ѫ���ر�Ե�Ǻ͵�������õIJ�λ�����Ƶ�С�ĵ���ͨ�������о���������ģ�� ��1998��Blodig��Doyle�ֱ�����LiP�����ϵľ���������ԭ���Ե�ɫ����л����ǵڶ����µġ���Ѫ���ر�Ե��Զ�̵���ת��λ�㲢ͨ����ѧ���κͶ���ͻ��չʾ���������λ�㡣Ŀǰϸ��ɫ��C��������ø��Ψһ��һ�ֱ�֤ʵ����ɫ����л����������ӵĹ�������ø����CCP�п���Ѫ����Я��ԭ���ŵ�ø�����︴���complex����ɫ����IJ�λ�ϣ�LiP��һ�����ܱ������ı�������л���EPR��ͼ��������LiP-I�д���߲����s�����ӣ�a porphyrin s-cation radial��������[9]��
1993��Piontek�Լ�Edwards���˷ֱ�����LiP�ľ���ṹ���������ṹ�Ĺ�ͬ���ʱ��LiP�����IJ�λ���ź�CcPһ�����۵��ṹ,Ѫ���ؿ�Ѩ��heme cavity���Լ����Ի��ŵ�����������dz����ơ���Ȼ��Ϊ����߾����ľ���������Ӵ���Ѫ���ص�[6]����Trp171λ����ʹ����������ķ�ʽ�γɣ���֤ʵ��Trp171 ��һ����������ԭ���Եİ����ᣬ����«�����������ڴ�ģ�͵Ļ����ϣ���«���������Ϳ��ܵ�ľ�ʻ���ص�Trp171��Ѫ���صĵ�����ͨ���ѱ��۲쵽�� �ڿ��ŵ�ͨ������Ѫ���صı�Ե��ͨ���Աߣ����Խ��С�ķ��������ӵIJ�λ������һ��С��ͨ�����Ե���Զ�˵�����Ѫ���ؿڴ��������ù���������ӽ��룩�����ҷ�����LiP�е�Ѫ���������Ӻͽ��˵��鰱��л��ϵ�Nԭ�ӵļ������Ա���ϸ��ɫ��C��������ø�ij�����LiP�н�����Fe-N�����ָ���ĵ��Ӵ��ݲ��ȶ��͵ĸ�����̬�����ԽϺõؽ���LiP��CcP�и��ߵ�������ԭ������LiP��һ���ӽ����ε���������ά�߶ȴ�Լ��50Å��40Å��40Å������Ѫ���طָ�Ϊ�����ֽ������Զ����Ѫ������ȫ��ֲ�뵽�����У�������ͨ��������С��ͨ���ӽ�������LiP������8����Ҫ��a������8��С������3���̵ķ�ƽ��a�۵�Ƭ��ÿһ������ֻ�������л���ɡ�Ѫ�������������и����ӽ�ϲ�λ��һ����Զ����һ���ڽ�������ԭ�ӵķ���������λ�ڻ���λ����鰱��л�֮��ͬʱ��7��8����ԭ����λ����ԭ��֮��ľ�����2.34��2.72Å[9]��
Ѫ���صĿ�Ѩ��cavity������40���л���ɣ�Ѫ�����ϵ���ԭ�Ӻ���ӽ���His176�ĵ�ԭ�ӽ�ϣ�����Ϊ2.15Å,������������ϵ�λ����һ��ˮ���ӣ���ͨ�������Զ�˵�His47�Լ�����������ˮ���ӽ�ϡ��ڱ�������Arg43��Ala180��Asn182��Asp183֮�䣬Ѫ����ͨ������͵���ϡ�Ѫ���غ͵���֮�䱣�����������Arg43��Asn182֮���γɵģ�������ӦҪ��һ�㣬�����3.20Å��EPR����ͼ����������ͼ��ʾѪ���ص���ԭ����ҪΪ��������������[9]��
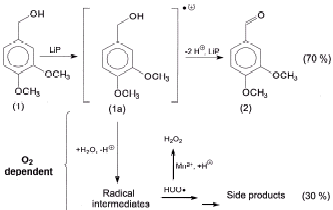
Haemmerli S.D.�������������ͼ3��ľ���ع�������ø����������ʱ������«��������ʾ��ͼ������Rimko ten Have�����ĺ��������о�ľ���ع�������ø����������ʱ������«���Ĺ�����������飬������Ϊ�ڷ�Ӧ�и�������γɲ���������ˮ��VA���ɻ������ӵ���������ɵģ�����Ϊ���Ƚ��е���������VA���ɻ������ӵ����ã�Ȼ�����ˮ�ġ���������
 |
ͼ3
��VAD���Լ�ľ���ع�������ø��VA���������и������γɵķ�Ӧ������ʾ��ͼ��VA��«��;VAD��«ȩ��[10] Fig.3 A schematic representation of the reaction mechanism involved in the formation of VAD and side products by the LiP catalyzed oxidation of VA |
ͼ
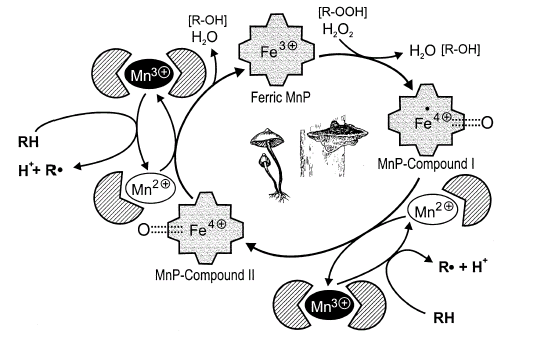
4. �̹�������ø�Ĵ�ѭ��ͼFig. 4 The catalytic cycle of manganese peroxidase (MnP)[11] 1.5 �̹�������ø(MnP,Manganese peroxidase)
���б��������ֲ�ͬ��MnP�̹�������ø�Ѿ����Դ���������һ���Ѿ��õ������С�ͨ����Ӿ������MnP�ķ�����Ϊ43Kda����ȵ����3.75������ø��һ�ֺ�5�����ǻ����ǵ��ס����������Ϊ43Kda���㣬���ְ�����л�����Ŀ�ǣ�50Asp��15Thr��49Ser��37Glu��33Gly��36Ala��17Val��4Met��11Ile��17Leu��7Tyr��21Phe��6His��5Lys��11Arg��19Pro��Cys��Trp��Ŀ��ȷ������
Mnp������ͬ��øͨ��SDS-PAGE��Ӿ���Ϊ44KDa����N��ȥ�ǻ��������У�MnP-I�ķ�����������42.7Kda����ʾ����3����N���ӵ����ࡣMnP1�ĵȵ����3.45��MnP2�ĵȵ����3.35��ͨ��IEF��ã�������LiP�ķ�������48Kda��n��ȥ�ǻ�������Ϊ45.5Kda��������5����N���ӵ����ࡣȥ�ǻ���������ͬ��ø�ĵȵ��ֱ���3.1��3.0�����������ʾ��Դ�� B. adusta �Ĺ�������ø������л��������(MnP1, LiP2): Asx (49, 56), Thr (26, 41), Ser(30, 19), Glx (39, 39), Gly (37, 39), Ala (47, 60), Cys(6, 6), Val (24, 22), Met (6, 10), Ile (19, 21), Leu (27,30), Phe (28, 36), His (9, 9), Lys (8, 7), Arg (10, 11),Pro (35, 35) and Tyr (0, 0) (Trp ��û�еõ�ȷ��)���� LiP �� MnP��ͬ��ø֮��û�з���ʲôϸ������[10]��
�̹�������ø�����й�������ø�ж��ص�һ��,���������Ļ�ԭ�����ΪMn2+�����MnP�Ļ�����I����������һϵ�з��㻯����,��������ľ�Ӻ�2,6-���������ӣ�2,6-dimethoxyphenol��, (Whitwam et al.,1997)��Ҫ��Mn2+��Ч�ʵ�,������II�Ļ�ԭ��ҪMn2+��Ϊ����.���Ŀ��ܵķ�Ӧ�����ǣ�
Ferric enzyme+H2O2��Compound I+H2O (1)
Compound I+RH��Compound II+R· ��2��
CompoundII+RH��Ferric enzyme+R·+H2O ��3��
Compound II+RH��Compound II--RH��Ferric enzyme+R·+H2O��4��
Compound II+H2O2��Compound III (5)
RH�ǻ�ԭ����,R*�ǵ�����������Ļ�ԭ����[12]
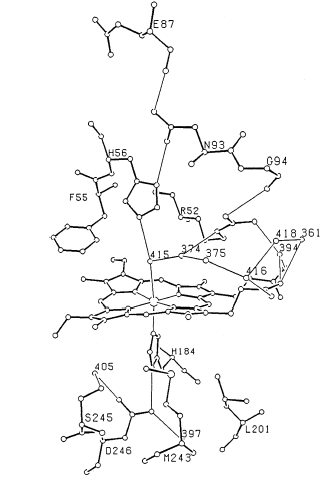 |
ͼ5 ARP��Ѫ���ػ��� Fig.5 Heme environment of ARP |
1.6 ARP(A.ramosus peroxidase)�Ĵ�����
��������Ѫ���صĹ�������ø������His�л���һ��Arg�л������������еĹ�������ø�ж��Dz���ġ����е��о������ʾ�ںܶ�Ĺ�������ø��һ��His�л��ڽ��˺�Ѫ�����е���ԭ����λ���ӣ���һ�����Ǻ�Arg�л�һ����Զ�˺���ԭ����λ������ѧ�о�������ARP(the A. ramosus peoxidase)��His 184��His 56�ֱ��Ӧ�ڽ��˺�Զ�˵�His, Arg 52��Ӧ��Զ�˵�Arg ��������о�����Ѫ���صĵ�����Эͬ����λ���ǰ����ӣ�pHΪ7.5ʱ�����á�415����ʾ��Toru Nakayama�������������ڽᾧ��ʱ��ʹ�ð�������ɵģ���ͼ5��ʾ��
�н����ʾARP�Ĵ���Ӧ�����ǰ������µIJ�����еģ�
ARP[Fe(III)]+H2O2��compound I[Fe(IV) ]+H2O (1)
Compound I[Fe(IV) ]+AH2��compound II[Fe(IV)]+AH (2)
compound II[Fe(IV)]+AH2��ARP[Fe(III)]+AH +H2O (3)
2AH ��A+AH2(��A2H2) (4)
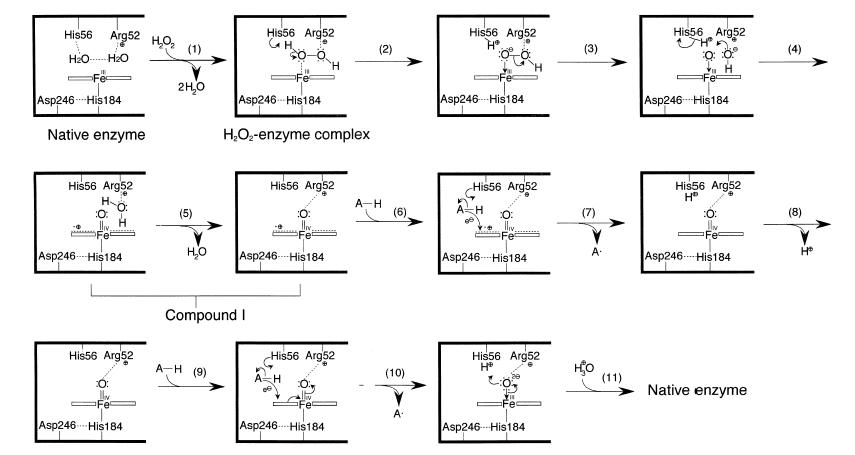
ͼ6 Fukuyama��Abelskov�����Ʋ��ARP�Ĵ�������������1��4��ʾ������I���γɣ�6��11��ʾ������I��II�����ι��̣�����5��Ϊ�����������ֶ������ġ����߱�ʾARP���ף�ƽ����α�ʾѪ����ƽ�棻���߱�ʾ����������ڵ�������á���������ø�Ĺ�ѧ���ݱ���������I����һ����������ɵ�߲������Fe����O����[14].
Fig.6 Proposed mechanism of ARP catalysis as depicted by Fukuyama ��Abelskov et al.. Steps 1 �C 4 and 5 for reductions of compounds I and II , steps 6 �C 11 with modifications. To tie both proposed mechanisms, step 5 has been introduced in this scheme. The bold line represents the ARP protein and the horizontal open rectangles indicate the heme plane. The broken lines indicate weak interactions including hydrogen bonding. Spectral studies of peroxidases show that compound I contains an oxygenated Fe IV with the cation radical.
ͼ
6����ʾ�Ļ�����I������1-4�ǽ����ԣ�ARP-I3����������Ľᾧѧ�о��Ļ����ϵġ��ڴ������ϣ�ARP��HRP�ļ�Ϊ���ơ�Զ�˵�Ѫ�����ṩ�˹�������ø�Ĵ����Բ�λ�����ص�His��Arg�л��ڴ��Ĺ��������˺���Ҫ�����á�����,ARP�Ļ�����I��II���ȶ��Ժ�pHֵ�Լ�����Һ��Ũ���кܴ�Ĺ�ϵ����ARP�л�����Iͨ������;�������������²���ԭø��native peroxidase������Ԥʾ����ARP�к�ϸ��ɫ��c��������ø��HRPһ����������I�ͻ�����II�������ƵĽṹ��������ARP�У�������I�ͻ�����II����廹ԭ���ܷ�����Ѫ���ر�Ե��Fe��O�Ǻ�Զ�˵�ɫ������ͨ�������ϵġ�
1.7 ����
Andrew T. Smith�����������н����е�ֲ�ﳬ�����й�������ø�Ĵ���Ӧ����һ������(��1)��ָ����Ȼ�����еķ�Ӧ��ø���ǽ�H2O2ת��ΪH2O�������ܺ������ǹ㷺�ز���������Ӧ�����Ҵֶ���������ѧ�����Եġ�����Ʋ����Ӧ����IJ�ͬ��ɵIJ���IJ�𣬽�����ԭ�ĵ��������Sred��ʾ����Sox��ʾ��������Ӷ��õ�һ��ֲ�ﳬ�����������ø����H2O2�μӵķ�Ӧ����ͨʽ����1��E��ʾ��������ø,Fe��ʾø�е�Ѫ���أ�(1)��ʾø�������ⷴӦ���ɻ�����I�������ӵļ�̬��+3�۱�Ϊ+4�ۣ�(2)������I�͵������õõ���������ͻ�����II��(3)�����ֺͻ�����II�����������������ͬʱ����ˮ��øҲ��Ϊ��ԭ����õ���ԭ��
��
1 ֲ�ﳬ�����������ø�Ĵ���Ӧ����[15]Table.1 The catalytic reaction mechanism of plant superperoxidases
E(FeIII,R)+H2O2
��E(FeIV=O,R·+) [Compound I]+H2O (1)E(FeIV=O,R·+)+Sred��E(FeIV=O,R)[Compound II]+Sox (2)
E(FeIV=O,R)+Sred��E(FeIII,R)+H2O+Sox (3)
L.DE Gioia���˱�������Ѫ���ع�������ø�������Ǵ����ֲ���������øһ�㶼��ѭͼ7�еĴ�ѭ������[16]��Gilda H.Loew���Թ�������ø�Ľṹ�����������ӻ�ѧ���㣬�õ���һЩ��������ø�Ĺ����������������ݣ�������Щ�����ƶ���ø�Ĺ������Ǵӷ�������ѧ�����ﻯѧ������ѧ������ѧ�ȸ�������Թ�������ø���˴������о�������Ҳ���һЩ��������ø�����һЩ����ģ�͡�
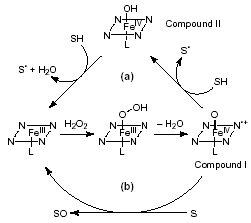 |
ͼ7 Ѫ���ع�������ø�Ĵ�ѭ��ͼ(a)Ϊ��������������(b)Ϊ˫������������ Fig.7 Catalytic cycles of heme peroxidases:(a) one-electron oxidation classical peroxidase reaction;and (b) twoelectron oxidation (oxygen transfer).Abbreviations: S, SH,substrate; SO, oxidized substrate. |
�Դ�1811��Thenard���ֹ�������ø����ָ����������Ի������ض��ĵ�����н����������ø���о�������Ļ�����Ƕ����Ĵ����ܡ��ṹ�����������Ļ������ģ��Լ����Ļ��������˺ܶ��о���ʹ���Ƕ�������ʶ�����ʵ���ߡ�������о��ֶ����ޣ�������Ҫ����������ѧ���о�����չҲ�����ޡ����ż����ֶε���ߣ���1936��Stern֤ʵ������ԭ߲��IX�ǻ��Ի��š�1937�꣬ţ�ι�������ø�ľ����Ʊ�������Ϊ���Ǵӽṹ����ʶ��������ø���������ṩ���о���������֮�����ǿ�ʼ�Ӿ���ѧ������ѧ����������ѧ���Ŵ�ѧ�ȸ��ֽǶȶԹ�������ø��չ����������о���
2.1��ˮ��ϵ���о�
�ڷ�ˮ��ϵ���Ծ��д���������ˮ������ø����Ӧ����ˮ��Һ�������߱����ŵ� ,��ø�����ȶ�����������ߵȡ��л�����ø�����������ۺ����й�ҵӦ��ǰ�����л���ø����Ӧ ,���Ƶ�������������֬���� ;���ȩ���ϳɵķ�ȩ��֬��� ,ø���ϳɵķ���֬���нṹ��һ��������ǿ���͵�����ص� ,������������������Ϳ�ϡ���¼���ϵ� ,�Ҳ������Ⱦ��Akkara��Dordick���о��˷�ˮ�����й�������ø������ۺ� ,�õ��˽ϸ���Է��������ķ���֬ ;���� ,�ۺ�ʱ������øʧ��Ͽ������ ,ø��ʹ�����ڽ϶� ,�Ӷ�����Ӱ����ø���ϳɵķ��������ϵijɱ��;ۺ϶ȵ���ߡ��������Ϻ��������Ľ�̫�ɵ����о���PEG���ε�������������ø�����ڷ�ˮ�����е�����[5] ��ͨ��������������ø (HRP)�����������백�������Ӽ��������Ҵ� (PEG5000)����ø������������ PEG5000 ,���� HRP�����������κ��ø��ˮ���еĻ�������û�б仯 ,��ͨ�������������� PEG��ø��ˮ���еĻ����½��� 2�����ڼױ����������������ϸߵ���ϵ�� ,����ø���������������ơ��ر��ڼױ���ϵ����������ø��������δ�����ε�ø����˽� 2�����ȶ����о����� ,������ˮ���л������л��� (����������ױ�)�� ,ֱ���� HRP������������ PEG5000��ʹø���ȶ����������ӡ������������� PEG��ø��ˮ���е��ȶ��Ա�û�����ε�ø�߶����л��� (�ױ����������)�е��ȶ��Ա�û�����ε�ø��[5]��
2.2��ѧ���⼼���ϵ�Ӧ��
�ڻ�ѧ���⼼���Ϲ�������øҲ������Ӧ�ã���HRP����һ�־�������Ŵ����õĹ�������ø,��ͨ������ɫ�������ʹ�ã�����ø�����������ⶨ (ELISA)������ҽѧ���顣����Ƽ���� ������CPD(��ѧ��������Ӱ��ⷨ)������ ,��ѧ����ʹX�⽺Ƭ�й���Ӱ�����Ը��ݽ�Ƭ�ϲ����ĺڰߴ�С����ɫŨ�����а붨���������ü����ⶨHRP�ļ����Ϊ 0.08�� 0.4pg(�� 2�� 10-18�� 2�� 10-17 mol) ,�Ȱߵ���ɫ�������ȸ� 50��500�� ,��ƽ��ELISA�����ȸ�700�� 1600������HRP��TAP���ε缫��Ӧ���ϣ�Annika Lindgren���˻�������һ���ڵ缫�Ͻ�øһ���̶��ķ���������Ƽ������ʵ��������ĵ�����Һ�����ﵽӦ��Ҫ�� ,�ⶨ������������ø (HRP)�������ȿɴﵽ 10-18 mol������ ,���ڲⶨDNA,������� 1pg�������ַ��ֲ�ͬ�ķ�����ǿ��ͬʱʹ��ʱ�Է�����Эͬ��ǿ���� ,�ɽ�һ����߲ⶨ�������ȡ����Ŵ�ѧ���ִ���ѧ�о�����1994�걨������HRPø���Ŵ���ϵ���������о�����,ʹHRP�ļ������ø������һ�������������Ծ��巴Ӧ��������̽�֡�
��������б����ӷ��������Elaeis guineensis����ȡ�õ��Ĺ�������ø�ڷ��⼼���ϵ�Ӧ�á����ֹ�������ø����������ͷ��ⰱ�ķ�Ӧ�����ʺ������õ���⡣���Ļ�ѧ����ǿ�����˵�pH��Χ�ǣ�8.3��8.6����һ��ATPOP��HRP��Arthromyces ramosus ��������ø��ʱ�����Ƶģ��̲ݹ�������ø����ʱ�������˵�pH��9.3���������Һ��Ũ�Ȼή�ͻ�ѧ����ǿ�ȡ�AOPTP �Ĵ�Ч�ʲ���������Ӧ��������ǿ��(4-��Ӻ�4-�ǻ������Ĵ��ڵ���һ�����Լ����ĸߵ��ȶ��Խ�����ʹ���ڴ��������ǰ;[17]��
2.3���ﴫ����
��������øҲ���Ա���������̽�롣������������̽���Ϲ̶����ø���ظ�ʹ�� ,�ȶ��Ժ� ,�����Ƽ��ͼ�����������Խ��� ,�ȽϾ��� ;�ټ���ø��������Ч��Ӧ�Ժ������ȣ�������������ø���ε缫�õ��˽�Ϊ�㷺���о��ͷ�չ������ H2O2 �����㰷�����ࡢ�����ǵ����ʵIJⶨ�����ź���Ҫ�����ã���Ϊø�缫�ṩ�˹�����Ӧ��ǰ����Wolenberger��Tatsuma�����Ʊ���HRP/������Ĥ�缫������˾����������á������ٽ�����Լ���������ï�� (FPF)��������������ø (HRP)���۽�� ,Ȼ��ͬ��������һ���ۺϵ����缫�� ,��ͨ�����ȩ������������ø (GOD)�����̶��ڵ缫�� ,�Ƴɹ������ε�HRP/GOD����ø�������������ʾ�������ε�ø��������Ӧ�������� ,������Է�Χ 0�� 40mmol/L ,�缫�ȶ�����ǿ��
���Ŵ�ѧ�������������ѧʵ������������Һ�������ڱ�������ۺ�ʵ����ø�Ĺ̶���������˾��������ܽ�������⣬ΪHRP�ҵ��������塣�±��͵��˻������ǵ������н��ǰ�˵Ĺ�����������ø�ĵ绯ѧ�̶��������Ʊ�������������ø /���ڱ�����Ĥ (HRP/ PPD)�缫�ķ�������������̵Ķ���ѧ���ؽ�����̽�֡�����誵����о�����N-�������Ϊ���� ,ͨ��ţѪ��������ȩʹ����Ϊ��ϵ���̼�缫��ȥ��������������ø���ﴫ��������ø�缫�Թ��������нϺõ���Ӧ��N-����ົ�ԭ��������ֵ���������Ũ���� 1�� 10-6�� 5�� 10-4 mol/ L��Χ�������õ����Թ�ϵ,�ô����������ȸ�,�����Ϊ10-7 mol/L,�Թ����������Ӧʱ��С��10s��
�����ױ����øõ缫��ⷼ�㰷�����������о���������չ�������˷��㰷�ܹ��ٽ�ֱ�Ӻͼ�ӵ�ø�缫��ԭ��Ӧ ,ø�缫��ԭ�����������뷼�㰷��Ũ����ء���������ڱ�ø�м��������� ,�ܹ��ڵ缫�ϵ绯ѧ��ԭ ,��ԭ��������Ũ�ȳɱ���[4,18]��
�����ڹ�ҵ���������������У�Ҫ��ˮ��Һ�� H2O2 �ĺ������м��;��HRP �кܸߵĴ�Ч�� ,ÿ��HRP������һ�����ڿ��Խ� 25000�� H2O2���Ӵ���ԭΪH2O����[20],��� HRP���ε缫��H2O2�ļ�����������ֱ�ӵ���ת�Ʒ�ʽ.��� H2O2ʱ ,ͨ��ѡ����Ƶ�λΪ 0 V(SCE),�ڴ˵�λ�� ,�������������ʵĸ��Ŷ���С ,����ʱ�߷�Ӧ�Ե�ø�м��� (������ - I /������ -II )����ͨ��ø��Ӧ������� ;�ü�ӵ���ת�Ʒ�ʽʱ ,�缫��λͨ��ѡ��ӽ����ӹ���ı���λ��ֱ�Ӻͼ�Ӳ���H2O2�ļ�����ɴﵽ 1.0�� 10-8mol/L����H2O2��˵ ,�缫�������ǹ̶�������HRP���Ǽ������ε� HRP,ø����Ӧ�ı������ϳ���������� ,��˵���������Ӱ��ø�Ĵ����ԡ����ò�ͬ�������ø���������߷�Ӧ���Ժ�ѡ���� [20]��
Ruzgas���� HRP���ε�̼���缫����ʯī�缫����˱��Ӽ���ص�һЩ�����廯���� ;Lindgren���ð��� HRP���ڵļ���ø�ֱ����ε�ʯī�缫�����һϵ�з������ ,�Լӵļ���ﵽ��10-7 mol/l��Dominguez-Sanchez������ HRP���ε�̼���缫��ⱽ�� ��HRP���ε缫�� H2O2�ļ���кܸߵ���Ч�� ,���Է��㰷���������ļ����ȱ��ѡ���� ��ø�缫Ŀǰ���������ڷӺͷ��㰷������IJⶨ ,��ø�缫��Һ��ɫ������ʹ�û����ܹ�����������[20] �������ԶԱ����ӵ��л���Ϊ���о��л�����ڶ� HRP/ PPDøĤ�缫�� H2 O2 ��ԭ�ٶȵ�Ӱ�� ,��������θ��ݶ���ѧʵ����ȷ���л�����ĺ���������Kulys����������������������ø (HRP)���ε�ʯī�缫��ijЩ�л�����вⶨ������̽�����䶯��ѧ��.ʵ���������Һ�д��ڱ���Ϊ����� (activator)�ķ�����ʱ ��HRPø�缫�� H2O2 ��ԭ������������
����л���������Ҳ�ǽ������о���һ���ȵ㡣����HRP���ε缫����л����������ԭ����HRP�� ROOH������η�����������Ӧ�����������ж�α����������о�������HRP���ε缫�� H2O2 ��Ӧ�Ľ��ͻ���������Һ�����Ƽ���������ж�Ӧ��ϵ[19]���ݴ˿ɼ�� HRP���Ƽ��ͼ���������������ⷽ�涼���˺ܶ����Bogdanovskaya���� HRP���ε�̼�ڵ缫��� HRP���Ƽ������� ,�����Ϊ 2.0�� 10 -4 mol· L- 1 [21];Smit���Զ�ï��Ϊý���� ,��� HRP���Ƽ��軯�� ,����ɴ� ng· m L- 1 ����Smit�Ȼ������̶�HRP�ļ������ü������ ,�ɼ� 5�� 10 -7 mol· L- 1 ����[22��23]�����ڽ����������Ƽ��ļ��,�������������Ǽ���Ϊ���ʵ� HRP���ε缫��� Hg2 +,����ɴﵽ pg· m L-1����Wang J�����л����� ,�� HRP���ε缫����˺ü��ֲ�������ˮ�� HRP���Ƽ������� ,HRP������ļ��Ҳ���о�[18��20]���������л��ܼ�/ˮ����������ʵIJⶨ�Լ�Ĥ�缫��ø�缫Ҳ��ʾ�˺ܺõ��ȶ��Ժ�ѡ���ԡ�
2.4����
�ڹ�ȷ����У�HRP����������Ҫ������ �������������� ,���������������Ⱦ�ϵ���ɫ��,������HRP���±�H2O2 ����Ϊ��ʽ�ṹ����ɫ������ӫ�������,���Dz�������HRP����Ӧ�ⶨ������Ʒ����H2O2����������ø��ż����Ӧ��ϵ�е�ָʾ��Ӧ ,���Ӽ�����������ԭø����ø��һЩ����ĺ����ȵȡ����ֿ��١�רһ���ϸ������ȵļ�ⷽ����ӫ������о�����Ҫ���塣�Ž��ơ�Mohanty JG��ǰ�˵Ĺ���������HRP����ͬ���������ӫ��������ǿ���֣���ø����Ӧ��ʵ�ʽ���̽�ֲ��ṩ��һ���µ�˼���Ƕȡ�
��������ø�ڻ��������Ӧ��Ҳ�Ǵ�������ĵ��ȵ�֮һ����������������۷��档��ǰ�ڳ������о��϶���������ͻ�ѧ�������ֶΣ��������ڴ�����Ⱦ��Ĺ����ж�ȱ���ϸߵĵ�ѡ���ԡ��������Ƿ���������Phanerochaete chrysosporium�е�ľ���ع�������ø�������Խ���ܶ�����Ⱦ�����DDT,PAH,PCB������LiP�����ǻ��о���������Ư�����к�ҵ��ˮ��LiP�Ը��־ۺ���Ⱦ�ϵĽ��⡣��������ż��Ⱦ������ɫƷ����Ϊ�ḻ��һ�࣬���Զ������о�Ҳ�����ġ�Paszczynski A���˷��ְ���Phanerochaete chrysosporium�е�ø����������ȥż��Ⱦ���е���ɫ�����Խ�����ת��ΪCO2��Helena Podgornik���˽�һ���о������ǻ���24��Ⱦ�����˲ⶨ������������Phanerochaete chrysosporium�е�ľ���ع�������ø���Զ����Բ�ͬ��ѧ�ṹ�������Ⱦ����ɫ�������߱������ԣ������õĵ��ﷶΧ�dz���[24]��Sami Sayadi�����ڶ���魼ӹ����ķ�ˮ�����ﴦ��ʱ���֣�LiP�����еĴ��к��Ķ��������Žϸߵ���ɫ�ͽ�����á�Monika Wagner���˷�����H2O2��������HRP�������Դ�����ţƤֽ�����������ŷŵĺ��������ʵĺ�����<1mg/L�������Խ���������Ķ��Զ��46���� 3. ������չ��
���ǽ�Ͻṹ�Զ��ֹ�������ø�Ĵ����̡��������Լ�Ӧ�õȷ������˺ܶ��о���������������������ø��ľ���ع�������ø���о�����Ϊ��ϸ������֮�⣬�����Ĺ�������ø�ķ�Ӧ���������Ǵ�ʵ�����õĽǶȳ������о��ϻ�δ�õ���ֺ�ϵͳ���о������������о��з��ָ������Ӷ�ø�Ļ����в�ͬ��Ӱ��,�Լ�����Ҳ�б�����������øֻ���������ӵļ�̬��+3�۵�ʱ��ž��д����õȵȣ�����Ҫ��ø�Ľṹ����ȥ�о����ǵķ�Ӧ�������������õ��������ǡ�
��������ø��һ���ȶ��ԣ�������pH�������ȶ��Եȵȣ��ܺõ�ø����������һ��������������ſ�ѧ��������ߣ��о��ֶεĸĽ������ǿ��Խ������ǵĽṹ�ͷ�Ӧ���������õ�Ӧ���ڹ�ҵ�����Ϳ�ѧ�о��� REFERENCES
[1] Marc B, Marc A. J. Am. Chem. Soc., 1997, 119: 576.
[2] Liu Junhong, Yang Fengke. J. Polymer Materials Science and Engineerin (Gaofenzi cailiao yu gongcheng), 2001, 17: 173-175.
[3] Haesun K, Harold E. Biochem., 1989, 28: 5714.
[4] Gaspara Szilveszter. Sensors and Actuators B, 2001, 72: 63-68.
[5] Jiang Taijiao, Jie Xingsong. Acta Biochemic et Biophysic Sinica (Shengwuhuaxue yu shengwuwuli xuebao), 1998, 30: 132-133.
[6] O'Brien, Peter J. Chemico-Biological Interactions, 2000, 129: 113-139.
[7] Gazarian Irina G. Biophysical Chemistry, 1998, 72: 231-237.
[8] Gazaryan Inna G, Lagrimini L Mark, Ashby Gilian A. J.Biochem., 1996, 313: 841-847.
[9] Choinowski Thomas, Blodig Wolfgang. J.Mol.Biol., 1999, 286: 809-827.
[10] Ten Have Rimko, Maurice C R Franssen. FEBS Letters., 2001, 487: 313-317.
[11] Hofrichter Martin. Enzyme and Microbial Technology., 2002, 30, 454-466.
[12] Heinfling Annaette, JesuE S MarEa. FEMS Microbiology letters., 1998, 165: 43-50.
[13] Mester Tuende, Tie Ming. International Biodeterioration & Biodegradation, 2000, 46: 51-59.
[14] Nakayama Toru, Amachi Teruo. Journal of Molecular Catalysis B: Enzymatic, 1999, 6: 185-198.
[15] Smith Andrew T, Veitcht Nigel C. Curr. Opin.Chem. Biol., 1998, 2 (2): 269-278.
[16] De Velde Fred van, Rantwijk Fred van, Sheldon Roger A. Trends in Biotechnology, 2001, 19 (2): 73-80.
[17] Sakharov I Y. Biochemistry (Moscow), 2001, 66 (5): 515-519.
[18] Ruan C, Yang F, Lei C. J. Anal. Chem., 1998, 70: 1721.
[19] Liu Y, Qian J, Fu X. J. Enzyme Microb.Technol., 1997, 21: 154.
[20] Jiao Kui, Zhang Shukui. Journal of Qingdao Institute of Chemical Technology (Qindao huagongxueyuan xuebao), 1999, 4: 333.
[21] Bogdanovskaya V A, Fridm an V A, Tarasevich M R et al. J. Anal. Lett., 1994, 27: 2823.
[22] Smit M H, Cass A E G. J. Anal. Chem., 1990, 62: 2429.
[23] Smit M H, Rechnitz G A. J. Anal. Chem., 1992, 64: 245.
[24] Podgomik Helena, Grgic Irena, Perdih Anton. Chemisphere, 1999, 38: 1353-1359. ��