http://www.chemistrymag.org/cji/2003/052016pe.htm |
Jan. 1, 2003 Vol.5 No.2 P.16 Copyright |
A density functional theory study of NO adsorption on the MgO (001) surface with various vacancies
Xu Yijun, Chen Wenkai, Li Junqian, Lu Naixia
(Department of Chemistry, Fuzhou University, Fuzhou, 350002, China; State Key Laboratory of Structural Chemistry, Fuzhou, Fujian,
350002, China)
Received on Oct. 8, 2002; Supported by the Natural Science Foundation of China (29973006)
and Administration of Science and Technology of Fujian Province (2001J018)
Abstract The adsorption of NO on the perfect and defective MgO (001)
surfaces has been studied using cluster models embedded in point charges with the
DFT/B3LYP method. The calculated results indicate that neutral oxygen vacancy (F center),
and charged oxygen vacancies (F+ center and F2+ center) are the
highly reactive catalysis structures for NO adsorptive-decomposition. The N-O bond length
was elongated much, which reflects that the NO is prone to decompose after adsorption on
the MgO (001) surface with oxygen vacancies. The N-O bond would be broken after an added
NO molecule approaches the adsorbed NO, along with a N2O molecule produced
simultaneously. This mechanism is consistent with the UPS and MIES experimental
observations. In contrast, neutral Mg vacancy (V center) and charged Mg vacancies (V-
center and V2- center) exhibit almost no catalytic reactivity for NO
decomposition on the surface, although there are very large adsorption energies.
Keywords NO, adsorption, MgO (001) surface, vacancies, DFT/B3LYP
1 INTRODUCTION
The intriguing heterogeneous processes associated with nitric monoxide, NO, observed at
metal as well as metal oxide surfaces, are a continuous topic for research [1].
NO, spontaneously produced in combustion processed at high temperature, is highly toxic
and has to be removed from the car exhaust gases. Therefore, it is necessary to develop
novel catalyst active in reduction of nitric oxides. In fact, numerous experimental
investigations and theoretical calculations have been performed regarding the adsorption
of NO on metal and metal oxides [2-10]. Among them, MgO, a typical alkali metal
oxide catalyst, is by far the most studied system. On a flat defect-free MgO (001)
surface, NO interacts very weakly with the surface [2,3]. On the other hand, IR
spectroscopy studies [5,6] of NO adsorption on preheated MgO surfaces have
proved that a variety of different surface species are formed. Some observed frequency
bands have been assigned to NO-, NO22-, NO3-
and N2O22- species, while other bands could not be
assigned. Some bands appeared as transients only. This rich diversity was assumed to arise
form the fact that the surfaces include not only the regular five-coordinated sites, but
also various sites displaying lower coordination numbers. The MIES and TPD experimental
data [7] showed that NO was dissociative adsorption at defect sites of MgO
(001) surface with concurrent production of N2O.Theoretical studies published [8-10]
about NO adsorption on the MgO (001) surface showed that several species can form on the
MgO surface upon adsorption of the NO radical. This depends on NO coverage, temperature,
and also on sample preparation and number of defects or impurities present on the surface.
Actually, the experiments and theoretical studies [11-14] have proved that the
MgO (001) surface with vacancies is more reactive than regular surface. However, so far,
systematic theoretical study of NO on MgO (001) surface with F, F+, F2+,
V, V-, V2- centers is scarce.
As such, it is very interesting and mandatory to understand the
mechanism of NO at vacancy sites of MgO (001) surface at the molecular level. This is also
the basis for further investigation of the reduction and decomposition of NO. For these
reasons, the possible models of NO on MgO (001) surface vacancy sites are proposed and
calculated in this paper. Besides, the interaction of NO on perfect surface is also
studied in order to compare its results with those on the surface with vacancies.
2 CALCULATION DETAILS AND MODELS
Solid MgO has the rock-salt structure with the lattice parameter 0.2014nm. Among the
various vacancies of MgO, oxygen vacancies and magnesium vacancies are common defects [13,15].
For the oxygen vacancy, according to the number of electrons trapped in the vacancy (2,1
or 0), this defect is called F, F+ or F2+ center. Similarly, for the
magnesium vacancy, the corresponding vacancy is named V, V- or V2-
center.
We used the Mg13O12 (Fig. 1 b) to simulate the
various oxygen vacancies (F, F+ and F2+ centers) of the MgO (001)
surface and the O13Mg12 (Fig. 1 c) for the magnesium vacancies (V, V-
or V2- centers), respectively. For the comparison to the MgO (001) surface with
various vacancies, the adsorption of NO adsorbed on the MgO (001) perfect surface was also
studied. High-resolution photoemission experiment proved that NO was bonded to metal
cations (Mg2+ sites) of MgO (001) perfect surface [3]. So, we only
used the Mg9O9 cluster (Fig 1. a) to calculate the adsorption of NO
at the Mg2+ site of the MgO (001) perfect surface. All these cluster models
were embedded in a large array of ±2.0 point charges(PCs) to take into account the
stabilizing effect of the rest of the crystal and, in particular, to ensure that the
Madelung potential in the chemisorption region was properly described [16,17].
The adsorption energies are defined positive for a stable
adsorbate/substrate system: Eads=ENO+EMgO-E(MgO+NO),
where E(MgO+NO) is the energy of adsorbate/substrate system in the equilibrium,
ENO and EMgO those of the adsorbate and substrate alone. Besides, we
have applied the standard counterpoise method [18] to account for the basis set
superposition errors (BSSE) for the calculations of adsorption energies.
For all the calculations, we used the hybrid density functional B3LYP [19,20]
method in Gaussian-98 program [21]. The DFT/B3LYP method is a three-parameter
hybrid density functional that combines the gradient-corrected exchange functional of
Becke and the correlation functional of Lee, Yang, and Parr with the Hartee-Fock exchange.
It is generally agreed that DFT methods give quite accurate results for solid systems [22].
The basis sets used in the present study are the standard 6-31g(d) for N, O and Mg.
Optimizations have been done by using Berny's
algorithm with Gaussian-98 program [21].
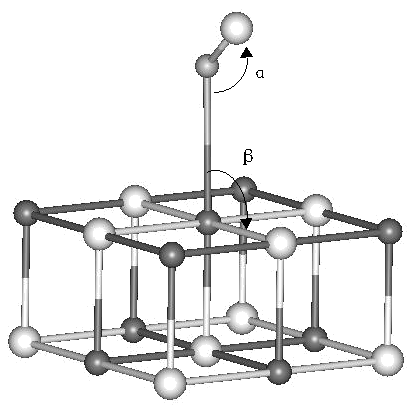 (a)
(a)
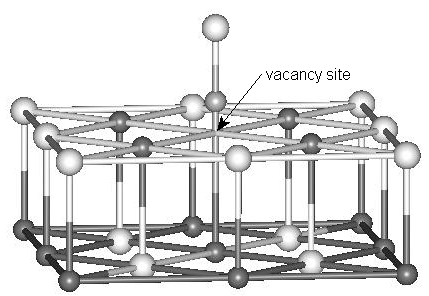 (b)
(b)
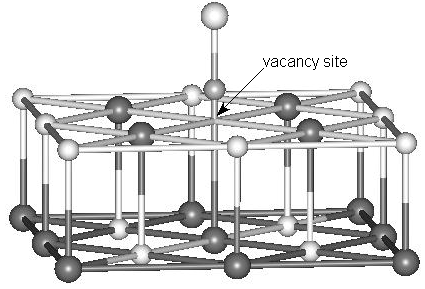 (c)
(c)
Fig. 1 Cluster models for NO adsorption at terrace and defect sites of the MgO
(001) surface. (a) NO adsorbed at the perfect site. (b) NO adsorbed at the oxygen vacancy
site (F, F+ or F2+ center). (c) NO adsorbed at the magnesium vacancy
site (V, V- or V2- center). Only the models with N end down on the
MgO surface are shown here. The surrounding point charges are not shown. Black spheres
represent Mg atoms, white spheres O atoms and grayish spheres N atoms.
3 RESULTS AND DISCUSSIONS
3.1 NO adsorption at perfect site of MgO (001) surface
For the free NO molecule, the N-O bond length and the dissociation energy calculated with
B3LYP method are 0.1159nm and 152.5kcal/mol, which are in good agreement with the
experimental values [8] of 0.1151nm and 150.1kal/mol, respectively. These
encourage us to make further investigations. High-resolution photoemission experiment [3]
has proved that NO is bonded to metal cations of MgO (001). Only the adsorption models of
NO on Mg site are calculated here. The adsorbed NO geometry is fully optimized. The
calculated geometry is shown in Table 1.
Table 1. The optimized results of NO adsorption on the MgO (001) surface
Cluster model |
R Mg-N(nm) |
R N-O(nm) |
a(°) |
b(°) |
Eads(kcal/mol) |
Mg9O9-NO |
0.2566 |
0.1153 |
131.22 |
91.43 |
5.69 |
Mg9O9-ON |
0.2614 |
0.1161 |
121.73 |
93.36 |
4.20 |
Free NO |
0.1159 |
From the adsorption
energy, we can clearly see that NO prefers to adsorb at the Mg site with N end down. The
calculated adsorption energy, 5.69kcal/mol, is lined with the average value of TDS
experiment [2] (5kcal/mol) and High-resolution photoemission experiment [3]
(4kcal/mol). The long distance of Mg-N, the undistorted intramolecular N-O bond length and
the low adsorption energy indicate that NO is weakly physically adsorbed on MgO (001)
surface. It can be concluded that the interaction of NO with the MgO surface is primarily
electrostatic interactions. The optimized angle of b, 91.43o, reflects that NO is prone to adsorb with N
vertically down on the MgO surface. Good consistency indicates that the DFT/B3LYP method
using embedded cluster models is suitable for the present study.
3.2 NO adsorption at vacancy sites of the MgO (001) surface
The calculated results for various adsorption models of NO adsorbed at vacancy sites of
the MgO (001) surface are shown in Table 2. Our interest here is to consider the effect of
vacancies, so we only consider the adsorption models of NO vertically down on the defect
surface.
Obviously, the adsorption energies of NO adsorbed at vacancy sites
increase much with respect to that on perfect surface. Energetically, NO prefers to adsorb
at the Mg vacancy site rather than the oxygen vacancy site. However, for NO adsorbed at
various Mg vacancy sites, the N-O bond length varies very little compared to the free N-O
bond. On the other hand, we can find that both large adsorption energy and large
elongation of the N-O bond length are found for the NO adsorbed at various oxygen vacancy
sites.
For NO adsorbed at the Mg vacancy site, NO is mainly adsorbed on the
surface with N end down. However, for the oxygen vacancy sites, both N and O end down on
the surface are possible. Moreover, it can be clearly seen that the N-O bond length,
besides the large adsorption energy, is elongated more when NO is adsorbed on the MgO
surface with O end down. This is particularly important in the case of NO, because in a
kinetic process it will not only increase the residence time of the molecule, but also
enhance the probability for dissociation. It can be deduced that the structure with oxygen
vacancies may have good catalysis structure for NO adsorptive-decomposition. Mulliken
charges analysis indicates that the electrons will transfer from the substrate to the
adsorbed NO and occupy the highest single electron occupied p* anti-bonding orbital for NO adsorbed at the oxygen vacancy sites,
which contributes to weaken the N-O bond. However, the case is not for NO adsorbed at the
magnesium vacancy sites, i.e. the electrons direction is from the p* anti-bonding orbital to the substrate
and strengthens the bond of NO to some extent.
Table. 2 The optimized parameters and Mulliken charges on NO for NO adsorbed at vacancy sites of MgO (001) surface
Cluster models |
R*X-W (nm) |
RN-O (nm) |
Eads (kcal/mol) |
e NO |
Mg13O12 (F center)-NO |
0.08114 |
0.1254 |
60.21 |
-0.417 |
| Mg13O12 (F center)-ON | 0.06836 |
0.1371 |
61.65 |
-0.444 |
Mg13O12 (F+ center)-NO |
0.08763 |
0.1237 |
30.43 |
-0.395 |
Mg13O12 (F+ center)-ON |
0.06563 |
0.1341 |
32.04 |
-0.448 |
Mg13O12 (F2+ center)-NO |
0.07644 |
0.1249 |
45.51 |
-0.393 |
Mg13O12 (F2+ center)-ON |
0.05699 |
0.1388 |
45.65 |
-0.447 |
O13Mg12 (V center)-NO |
0.05745 |
0.1120 |
72.81 |
0.431 |
O13Mg12 (V center)-ON |
0.1169 |
0.1099 |
53.41 |
0.584 |
O13Mg12 (V- center)-NO |
0.05476 |
0.1124 |
41.77 |
0.405 |
O13Mg12 (V- center)-ON |
0.1092 |
0.1103 |
17.74 |
0.531 |
O13Mg12 (V2- center)-NO |
0.05998 |
0.1125 |
64.79 |
0.401 |
O13Mg12 (V2- center)-ON |
0.08716 |
0.1098 |
32.38 |
0.584 |
Free NO |
0.1159 |
0.000 |
* X denotes the defect site (oxygen vacancies or magnesium vacancies). W denotes the adsorption site (N site or O site) of NO.
3.3 The mechanism of NO dissociation on the
MgO (001) defect surface
One possible mechanism of NO dissociation on the defect surface site is investigated.
Namely, NO will convert to N2O after another NO molecule approached the
adsorbed NO. At the same time, the O atom of adsorbed NO will become the lattice oxygen.
For simplicity, we only select the system Mg13O12 (F center)-ON to
study the possible mechanism of NO decomposition (see Fig. 2). The optimized geometry is
listed in Table 3.
Table 3. The optimized geometry of Mg13O12 (F center)-ON-NO system
Cluster model |
R1 (nm) |
R2 (nm) |
R3 (nm) |
R4 (nm) |
Mg13O12 (F center)-ON-NO |
0.00212 |
0.2879 |
0.1142 |
0.1177 |
Free N2O |
0.1134 |
0.1193 |
It can be seen that the
adsorbed N-O bond is almost broken when another NO attaches it because the R2
is long enough for the dissociation. The Mulliken overlap-population 0.0017 corresponding
to the R2 also supports this. At the same time, we can see that the optimized R3
and R4 are also close to those of the free N2O molecule. All these
indicate that NO is broken and N2O is concurrently produced. The decomposed
atomic O seems to become the lattice oxygen (see R1). The proposed mechanism is
consistent with the TDS and MIES experiments, which found that the NO would be
dissociative on defect sites of MgO (001) surface with concurrent production of N2O.
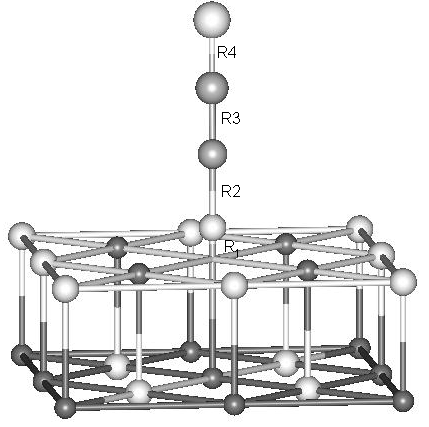
Fig. 2 The sketch of another NO molecule
approaching the adsorbed NO
For the dissociative
chemisorption of NO after another one attaches the adsorbed NO:
![]()
Where, X represents the defect neutral oxygen site of MgO
(001) surface. We calculate the energy released in this reaction is about 71kcal/mol.
Thus, the dissociation of the NO is a much energetically favorable process.
The calculated results indicate that the MgO (001) surface with oxygen
vacancy is a good candidate to catalyze the NO dissociation.
4. CONCLUSTIONS
The adsorption of NO on the MgO (001) surface with various oxygen vacancies (F, F+
and F2+ centers) and magnesium vacancies (V, V- and V2-
centers) has been investigated. The calculated results can be summarized into the
following: (1) Both the surface structure with oxygen vacancies and that with magnesium
vacancies are the good adsorption substrates for NO adsorption. The large adsorption
energies of NO on vacancy sites of the MgO (001) surface reflect that it is strongly
chemisorbed. (2) The surface with oxygen vacancies (F, F+ or F2+
center) is helpful to dissociation of NO, especially for NO adsorption at the surface
defect site with O end down. (3) A possible mechanism of NO conversion into N2O
is proposed, i.e. NO will decompose after another one approaches it. The optimized
structure indicates that this process seems to be natural because the adsorbed N-O bond
length is elongated significantly. The large released energy for the reaction of NO
dissociation also supports this.
REFERENCES
[1] Overbury S H, Mullins D R, Huntley D R et al. J.Catal., 1999, 186: 296.
[2] Freund H J. Faraday Discuss., 1999, 114: 1.
[3] Rodriguez J A, Jirsak T, Kim J Y et al. Chem.Phys.Lett., 2000, 330: 475.
[4] Sorescu D C, Rusu C N, Yates J T. J.Phys.Chem B, 2000, 104: 4408.
[5] Yanagisawa Y. Apply.Surf.Sci., 1996,100/101: 256.
[6] Platero E E, Spoto G, Zecchina A. J.Chem.Soc.,Faraday Trans. 1985, I81: 1283.
[7] Kolmakov A, Stultz J, Goodman D W. J.Chem.Phys., 2000, 113: 7564.
[8] Lu X, Xu X, Wang N Q et al. J.Phys.Chem B, 1999, 103: 5657.
[9] Yanagisawa Y, Kuramoto K, Yamabe S I. J.Phys.Chem B, 1999, 103: 11078.
[10] Lu X, Xu X, Wang N Q et al. Chem.Phys.Lett., 1999, 300: 109.
[11] Kantorovich L N. Surf.Sci., 1995, 343: 221.
[12] Coluccia S, Barton A. J.Chem.Soc.,Faraday Trans.I, 1981, 77: 2203.
[13] Sushko P V, Shluger A L, Catlow C.Richard A. Surf. Sci., 2000, 450: 153.
[14] Nada R, Hess A C, Pisani. Surf.Sci., 1995, 336: 353.
[15] Ferrari A M, Pacchioni G. J. Phys. Chem., 1995, 99: 17010.
[16] Pacchioni G. Surf.Sci., 1993, 281: 207.
[17] Pacchioni G, Clotet A, Ricart J M. Surf.Sci., 1994, 315: 337.
[18] Boys S, Bernardi F. Mol. Phys., 1970, 19: 553.
[19] Becke A D. J. Chem. Phys., 1993, 98:5648.
[20] Lee C, Yang W, Rarr R G. Phys. Rew. B, 1988, 37: 785.
[21] Frisch M J, Trucks G W, Schlegel H B et al. Gaussian 98. 2nd edition.
Pittsburgh PA: Gaussian Inc., 1998.
[22] Ziegler T. Chem. Rew., 1991, 91: 651.