http://www.chemistrymag.org/cji/2003/055036pe.htm |
May 1, 2003 Vol.5 No.5 P.36 Copyright |
Investigation of
nano-titanium oxide photocatalysts for the decomposition of nitrogen oxide in the flow
system
Zhang Jinlong, He Bin, Masakazu Anpo#
(Institute of Fine Chemicals, East China University of Science and Technology, Meilong
Road 130, Shanghai 200237, China; # Department of Applied Chemistry, Osaka
Prefecture University, Gakuen-cho, Sakai, Osaka 599-8531, Japan)
Received Dec. 10, 2002; Supported financially by the National Natural Science Foundation
of China (No.29476231), Foundation of Ministry of Education P.R.C., Shuguang Plan of
Commission of Education of Shanghai, and Nano-materials Special Item of Commission of
Shanghai Science and Technology.
Abstract In order to investigate the mechanism of the decline phenomena
of nano TiO2 photocatalysts in the photocatalytic decomposition of NO in a
large scale flow reaction system, the TPD (temperature-programmed desorption) spectra
adsorbed species at the photocatalysts were investigated in detail before and after the
photoreactions. It was found that the TPD peak assigned to the adsorbed NO species
observed at around 524 K is directly associated with the decline in the photocatalytic
reactivity of TiO2. The TPD spectrum of the adsorbed NO species consisted of
three different types of species. The g-adsorbed species of NO formed under UV irradiation on the active
surface sites cause a remarkable decline in the photocatalytic reactivity of TiO2.
It was also found that such a decline caused by the strong adsorption of NO at the surface
sites can be recovered to its original reactivity by heat treatment in atmosphere Ar
and/or O2 at around 573 K.
Keywords photocatalysis; titanium oxide; decomposition of NO; TPD spectra
1. INTRODUCTION
A great deal of attention has been aimed at the investigation of nano-photocatalysts which
could be applied for improvement of human life environment. Although this field has
expanded rapidly with regard to light energy, especially the conversion of solar energy
into useful chemical energy using solid photocatalysts such as TiO2,[1-4]
more recently, the application of photocatalysis to reduce toxic agents in the atmosphere
and water is being actively investigated. Nitrogen oxide(NOx) is an especially harmful
atmospheric pollutant which causes acid rain and photochemical smog. The direct
decomposition of NOx into N2 and O2, has been a great challenge for
many researchers.[4-11] TiO2 is especially attractive due to its
various properties as a non-toxic, highly stable, low cost, and high activity,
significantly, for chemical waste remediation. [12-16]
Up to date, there have been a few reports concerning the origin of the often observed
decline in the photocatalytic reactivity of TiO2 photocatalysts in both aqueous
and gas phase reaction systems.[16-24] The design and development of titanium
oxide photocatalysts for use in a large scale will only be possible by investigating and
understanding the various aspects of the reactivity including the origin and recovery of
this decline. In the present study, we have carried out observations of the decline in the
photocatalytic reactivity of titanium oxide photocatalysts for the decomposition of NO
under a large scale flow system and investigated the origin by means of a
temperature-programmed desorption (TPD) method. Moreover, we have investigated that how
the lost photocatalytic reactivity can be recovered to its original reactivity level.
2. EXPERIMENTAL
2.1 Photocatalytic flow reaction system
The photocatalytic reactions were carried out in the flow system described in the
preceding paper.[25] The flow rates of the reacting gases (NO, O2)
and pretreatment gases (Ar, O2) were adjusted by a mass flow controller. An
electronic furnace was used to pretreat the catalysts under an O2 and Ar flow
at suitable temperatures. During the photoreactions, to keep the Hg-lamp cool and to
absorb the infrared beams emitted, water was cycled in cooling pipes made of quartz glass.
A NOx meter (New Cosmos Electric Motors, Ltd.) and gas chromatography
were used to measure the NO and product (N2, O2 and N2O)
concentrations. The reaction cell was made of quartz (length: 190 mm; inner diameter: 10
mm). 150 mg of the TiO2 photocatalyst was introduced into the cell. To prevent
any movement of the catalyst in the cell, glass wool was placed at both the bottom and top
sides of the reaction cell. The NOx meter and gas chromatographer were connected to a
recorder which continuously measured changes in the concentrations of NO and the products
of N2, O2 and N2O.
2.2 Catalysts
Five different types of nano powdered TiO2 catalysts (grain size, 0.02-0.1 mm)
supplied by the Catalysis Society of Japan as standard reference TiO2
(JRC-TIO-1, 2, 3, 4, 5) were used as the photocatalysts.[25] Detailed
information on these standard reference TiO2 catalysts is available from the
Catalysis Society of Japan.
2.3 Photocatalytic Decomposition of NO
Photocatalytic reactions were carried out under a flow system.[25] The flow
reactant gases and the pretreatment gases were introduced from the bottom side of the
catalyst layers. 150 mg of the TiO2 photocatalyst was loaded into the reaction
cell which was then connected to the flow system with silicone grease. The TiO2
photocatalyst was heated in O2 using an electronic furnace, and then treated
with Ar flow gas at 20 cc/min. When the temperature of the photocatalyst reached the
suitable temperature, O2 and Ar flow gases were introduced into the cell for 2
h at 20 cc/min, respectively. The system was then cooled down to the reaction temperature,
the O2 flow was discontinued although the Ar flow was continued further for 1 h
at a flow rate of 20 cc/min. After these pretreatment proceduces, the reaction cell was
cooled down to room temperature under an Ar gas flow.
In the photocatalytic reaction, the concentration of the NO reactant
gas (mixture of NO and Helium) was 10 ppm, and the flow rate of the reactant gas was 100
cc/min. After reaching a steady state, UV irradiation was started and continued for 2 h. A
Toshiba SHL-100UV high-pressure Hg lamp was used. The distance of the lamp from the
reaction cell was 10 cm and a color filter of UV-27 (l> 270 nm) was used. The photon density of the Hg lamp passing
through the UV-27 filter was 10790 Lux, measured by a Digital Lux meter (model LX-1332).
2.4 TPD measurements
After the photocatalytic decomposition reaction of NO in the hermetic system, the TPD of
the TiO2 photocatalyst was measured. 10 mg of TiO2 was loaded into
the TPD cell. Before each run, the TiO2 photocatalyst was heated to 573 K at 5
K/min in 3 kPa (23 Torr) of O2 in order to create an original oxide surface,
and then evacuated for 2 h. After degassing the sample at 573 K for 1 h, the TiO2
sample was cooled to room temperature. Then after 1 kPa (7 Torr) of NO was introduced into
the TPD cell, the TiO2 sample was irradiated for 2 h by UV light. The TiO2
was degassed in the system for 15 min prior to TPD measurements. Temperature-programmed
desorption was measured by a M-QA100TS mass analysis meter equipped with a PC-9821
computer. During TPD measurements, the TiO2 catalyst temperature was increased
at a constant rate of 5K/min controlled by a Digital Programm Temperature Adjuster FP21.
3. RESULTS AND DISCUSSION
3. 1 The origin of the decline of the photocatalytic activity
Standard reference TiO2 photocatalysts JRC-TIO-1-5 were used in the flow system
to study the origin of the decline in efficiency during long time utilization. Figure 1
shows that JRC-TIO-4 gradually loses its photocatalytic reactivity during photoreactions
carried out on a large scale and for a long period. After 2 h, the conversion of NO for
each catalyst levels off and becomes 1/4-1/5 of the reactivity at the beginning of the
reaction. Thus, the decline in the photocatalytic reactivity of TiO2 for the
decomposition of NO in the absence of O2 and/or H2O could be
confirmed. The reaction products of the decomposition of NO in the flow system were also
confirmed to be N2, O2 and N2O, as with the closed
reaction system.
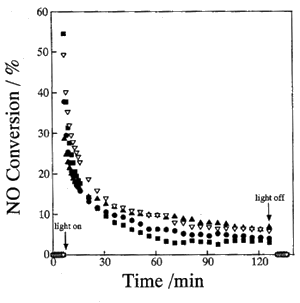
Fig.1 Reaction time profiles of the photocatalytic decomposition of NO at
room temperature on the TiO2 photocatalyst pretreated with O2 at 573
K.
Pretreatment: after introduction of O2 (20 cc/min) and Ar (20 cc/min) at 573 K,
heated in Ar flow (20 cc/min) at 295 K (��), 373 K (��), 473K (��) and 573 K (��), respectively. Gas component: 10 ppm NO, 100 cc/min. Catalyst:
JRC-TIO-4, 150 mg.
The adsorbed compounds cause the photocatalytic deactivation in the NO
decomposition reaction.[26] It was found that such a decline in reactivity
after the reaction of NO under a large scale flow system could be recovered to its
original level by applying heat treatment to the used catalysts under an O2 or
Ar flow at around 573 K for one h, as shown in Fig. 2.
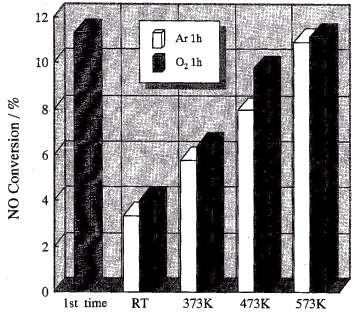
Fig. 2 NO conversion of the photocatalytic decomposition of NO on the TiO2
photocatalyst recovered by treatment under a flow of Ar or O2 for 1h at
different temperatures. Pretreatment: after introduction of O2 (20cc/min) and
Ar (20cc/min) at 573 K, heated in Ar flow (20cc/min) at 573 K. Gas component: 10 ppm NO,
100 cc/min. Catalyst: JRC-TIO-4, 150 mg.
In this treatment, the declined reactivity of the TiO2
photocatalyst could be greatly recovered by heating in Ar at 473 K.[27] After
Ar heat treatment at up to 573 K, the reactivity of the TiO2 photocatalyst
recovered almost to its original level. Thus, in order to address the observed decline, we
found that a complete recovery of the reactivity to its original level could be achieved
by heat treatment with Ar or even with O2 at 573 K. These results clearly
indicate that the decline phenomena of TiO2 for the decomposition of NO are
caused by the adsorption species on the surface inhibiting the subsequent reactions.
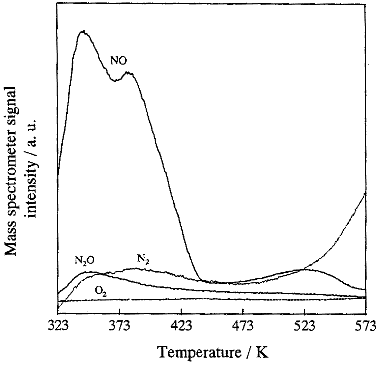
Fig.3 TPD profiles of the adsorbed NO, N2O, N2 and
O2 recorded after the photocatalytic decomposition of NO on JRC-TIO-4 at room
temperature. Catalyst: 10 mg. Vacuum pressure: 10-5-10-6 Pa (8x10-8
- 8x10-9 Torr). Heating rate: 5 K/min.
3.2 TPD measurements of the TiO2 photocatalysts after photocatalytic reactions
In order to investigate the kinds of adsorbed species which may be associated with the
decline in the photocatalytic reactivity of TiO2, temperature-programmed
desorption (TPD) measurements of the catalysts were carried out after the reactions. From
Fig. 3, it is clear that the decline is strongly associated with the adsorbed species on
the photocatalyst, and not due to changes in the surface structure of the photocatalyst,
since the photocatalytic reactivity can be recovered by Ar heat treatment at 573 K. As it
can be seen in Fig. 3, with the TPD spectrum, the desorption peaks assigned to N2,
N2O and NO can be observed. The chemical strectures of N2, N2O
and NO were confirmed by a mass analyzer, however, the desorption peaks assigned to O2
and NO2 cannot be detected. The appearance of peaks due to N2O and N2
in addition to NO clearly indicate the decomposition of the adsorbed NO. Therefore, it can
be confirmed that the decline phenomena is not caused by the adsorption of O2.
Furthermore, a single peak is observed at around 348 K due to the desorption of N2O,
and it was found that the adsorbed species of N2O is completely dissociated up
to 473 K. Figure 2 shows that the decline in photocatalytic reactivity could not be
recovered to its original level without heat treatment of the catalyst with Ar or O2
at 573 K. These results clearly indicate that the adsorbed species of N2O which
were completely decomposed into N2 and O2 at around 473 K are not
associated with the decline. The TPD spectrum assigned to N2 consists of two
different components, i. e., a peak at around 373 K and another peak higher than 573 K.
The peak at around 373 K is the same as that observed for the N2O species which
completely decomposed into N2 and O2 at up to 473 K, while the other
N2 peak is higher than 573 K showing the strong adsorption property of N2
which cannot be dissociated even with treatment at 573 K with Ar or O2. These
results clearly indicate that the N2 adsorbed species does not induce the
decline in the photocatalytic reactivity. In addition to these peaks, three peaks assigned
to the adsorbed NO species were observed at around 343 K, 381 K and 524 K. The
dissociative peaks at around 343 K and 381 K could be eliminated by treatment at 473 K,
however, the declined photocatalytic reactivity could not be recovered to its original
level. It is, therefore, possible that the dissociative peaks of NO at 343 K and 381 K
have little associated with the decline in photocatalytic reactivity. For the dissociation
of the adsorbed NO species at around 524 K, the TPD peak can be observed between 473 K and
573 K. With the original TiO2 photocatalyst, such desorption peaks could not be
observed. In fact, they could only be observed with TiO2 catalysts which showed
a decline in photocatalytic reactivity, as shown in Fig. 2. From these results, we can
conclude that the decline in the photocatalytic reactivity of TiO2 in the
decomposition of NO at 295 K is strongly associated with the formation of strong
adsorption species of NO on the TiO2 surface, which are perhaps able to adsorb
on the strong acidic sites working as active surface sites on the photocatalyst. These NO
species can be eliminated by treatment of the catalysts with Ar or O2 at
temperatures as high as 573 K.
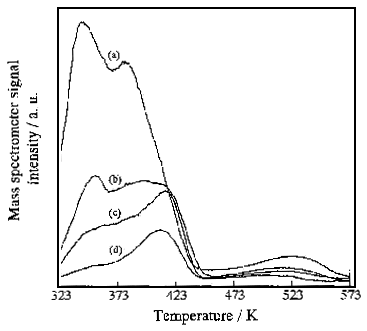
Fig. 4 Effect of UV irradiation time on the TPD profiles recorded after
the photocatalytic decomposition of NO. Irradiation time: (a) 120 min, (b) 30 min, (c) 10
min, (d) 0 min. Catalyst: 10 mg. Vacuum pressure: 10-5~10-6 Pa (8x10-8
~ 8x10-9 Torr). Heating rate: 5 K/min.
3. 3 The effect of UV irradiation on the TPD spectrum of NO
Figure 4 shows the changes in the NO TPD spectra with the irradiation time in the
photocatalytic decomposition of NO. The intensities of the TPD spectra become stronger
with the extending UV irradiation time. The deconvolutions of the NO TPD patterns are
shown in Fig. 5. The spectra consist of three different components, the a, b, and g species, each having a different dissociation temperature. The
deconvolution spectra recorded after the reactions under various UV irradiation times are
shown in Table 1, where it can be seen that the intensities of the a, b, and g TPD peaks remarkably increase with the UV irradiation time. In
particular, the intensity of the a TPD peak due to NO is greatly increased compared with
those of the b and g species. It is likely that the NO
adsorbed species assigned to the a peak plays a more important role in the photocatalytic
decomposition of NO than the b and g species. The TPD peak positions of the a,
b, and g species were found to shift and change with the UV irradiation
time. The peaks of the a, and
b species shifted to lower
temperature regions with prolonged UV irradiation. However, the peak of the g species which seems to play a vital
role in the decline of the photocatalytic reactivity of TiO2 for the
decomposition of NO shifted to higher temperature regions, in contrast to the a and b species. These results clearly indicate that the g-species which are formed by UV
irradiation adsorb strongly onto the surface of TiO2, i. e., on the
active sites for the photocatalytic decomposition reaction of NO, causing a remarkable
decline in the photocatalytic reactivity of TiO2 under UV irradiation.
Therefore, to develop highly active TiO2 photocatalysts, the surface of TiO2
must be modified to avoid and resist the strong NO adsorption properties of this species,
investigations of which are presently underway in our laboratory.
Table 1. The results of the deconvolution of the TPD spectra of TiO2
after the photocatalytic reaction for different UV irradiation time intervals.
UV irradiation |
Integrated area /a.u. | Desorption temperature / K | ||||
| a | b | g | a | b | g | |
| 0 | 1.0 | 1.0 | 1.0 | 371 | 411 | 498 |
| 10 | 2.4 | 2.0 | 1.1 | 360 | 411 | 511 |
| 30 | 3.9 | 3.0 | 2.6 | 354 | 407 | 512 |
| 120 | 7.8 | 5.0 | 4.3 | 346 | 393 | 516 |
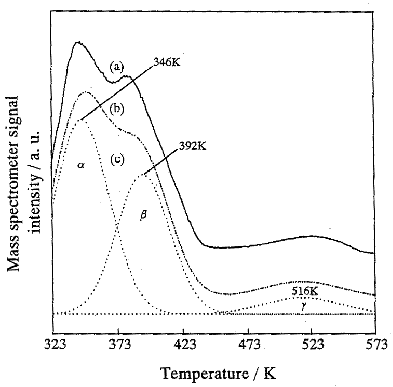
Fig. 5 TPD profiles recorded after the photocatalytic decomposition of
NO. (a): observed TPD profiles of a, b, and g shown in (c); (b): sum of the deconvoluted peaks shown in (c),
(c): deconvoluted peaks, a, b and g.
It has been reported that
the properties of adsorption and dissociation of NO are different on metals and oxides
when their crystal faces are different.[26, 27] Because there are differing
anatase and rutile constitutions in the standard reference catalysts JRC-TIO-1 ~
JRC-TIO-5, and their physicochemical properties are also different, these factors will
naturally affect the TPD patterns. The peak positions of NO, N2O, N2
and O2 on the standard reference catalysts used are shown in Table 2 along with
the differences observed in the dissociation species, dissociation temperatures and
numbers of TPD peaks when the physicochemical properties of TiO2 are different.
The TPD patterns of NO on the standard reference catalysts are shown in Fig. 6. From Fig.
6, NO peaks of lower temperature can be observed in all the standard reference catalysts,
and those peaks can be removed when the treatment temperatures are raised up to 473 K. The
intensities of the peaks of the dissociation NO species in JRC-TIO-3 and JRC-TIO-4 are
strong, showing that their reactivities are higher in the photocatalytic decomposition of
NO. It is possible to assign the peak at the lower temperature to the a species, i. e. the
adsorbed NO species assigned to the a peak plays a more important role in the
photocatalytic decomposition of NO than the b and g species. However, the direct relationship between the differences
in the anatase and rutile structures, surface area, surface -OH groups, band gap and their
dissociation patterns have not yet been concluded.
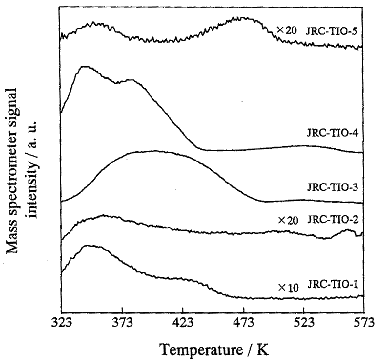
Fig.6 TPD profiles of the adsorbed NO species recorded after the
photocatalytic decomposition of NO at room temperature for the different TiO2
photocatalysts.
Table 2 The results of the analysis of the observed TPD patterns due to
the adsorbed NO, N2O, N2, and O2 on standard reference
TiO2 photocatalysts after the photocatalytic decomposition of NO at room
temperature.
| �� | �� | ||||
| �� | 1(anat.) | 2(anat.) | 3(ruti.) | 4(anat.) | 5(ruti) |
| NO | 349K 435K(sh) |
357K(br) |
401K 523K(br) |
343K 381K 524K(br) |
353K 475K |
| N2O | - | - | 365K | 348K(br) | 573K |
| N2 | 473K |
355K |
403K(br) |
380K(br) 573K< |
�� |
| O2 | 573K< | - | - | - | - |
sh=shoulder, br=broad
4 CONCLUSIONS
In order to investigate the origin of the decline in the photocatalytic reactivity of TiO2
in the decomposition of NO under a flow reaction system, the TPD spectra of the TiO2
photocatalysts were investigated in detail after the photoreactions. It was found that the
TPD peak assigned to the adsorbed NO species observed at around 524 K is directly
associated with the decline in the photocatalytic reactivity under UV irradiation. The TPD
spectrum of the adsorbed NO species consisted of three different NO adsorbed species of
which the g-adsorbed
species formed on the active surface sites under UV irradiation were found to cause the
drastic decline in the photocatalytic reactivity of TiO2. It was also found
that the recovery of the reactivity to its original level can be achieved by heat
treatment with Ar and/or O2 at around 573 K.
REFERENCES
[1] Anpo M, Yamashita H. in "Surface Photochemistry", Anpo, M., Ed., John Wiley
& Sons: Chichester, U. K., 1996, 117.
[2] Anpo M. Catal. Surv. Jpn., 1997, 1: 169.
[3] Hoffmann M R, Martin S T, Choi W et al. Chem. Rev., 1995, 95: 69.
[4] Fox M A, Dulay M T. Chem. Rev., 1993, 93: 341.
[5] Ollis D F, Al-Ekabi H Eds. Photocatalytic Purification and Treatment of Water
and Air. Elsevieer: Amsterdam, 1993.
[6] Ichihashi Y, Yamashita H, Anpo M. Study. Surf. Sci. Catal., 1997, 105: 1609.
[7] Anpo M, Ichihashi Y, Takeuchi M et al. Res. Chem. Intermed., 1998, 24 (2): 143.
[8] Serpone N, Pelizzetti E, Photocatalysis. Wiley, New York, 1989.
[9 Fan J, Yates J T. J. Am. Chem. Soc., 1996, 118: 4686.
[10] Lichtin N N, Avudaithai M. Environ. Sci. Technol., 1996, 30: 2014.
[11] Yamashita H, Zhang S G, Matsumura Y et al. Appl. Surf. Sci., 1997, 121&122: 305.
[12] Alekabi H, Serpone N. J. Phys. Chem., 1988, 92: 5726.
[13] Yamashita H, Honda M, Harada M et al. J. Phys. Chem. B, 1998, 102: 10707.
[14] Yamashita H, Kawasaki S, Ichihashi Y et al. J. Phys. Chem. B, 1998, 102: 5870.
[15] Nimlos M R, Jacoby W A, Blake D M et al. Environ. Sci. Technol., 1993, 27: 732.
[16] Park D R, Zhang J L, Ikeue K et al. J. Catal., 1999, 185: 114.
[17] Phillips L A, Raupp G B. J. Mol. Catal., 1992, 77: 297.
[18] Obee T N, Brown R T. Environ. Sci. Technol., 1995, 29: 1223.
[19] Larson S A, Falconer J L. Catal. Lett., 1997, 44: 57.
[20] Yamashita H, Ichihashi Y, Harada M et al. J. Catal., 1996, 158: 97.
[21] Peral J, Ollis D F. J. Catal., 1992, 136: 554.
[22] d'Hennezel O, Ollis D F. J. Catal., 1997, 167: 118.
[23] Cunningham J, Hodnett B K. J. Chem. Soc. Far. Trans., 1981, 77: 2777.
[24] Larson S A, Falconer J L. Catal. Lett., 1997, 44: 57.
[25] Zhang J L, Ayusawa T, Minagawa M et al. J. Catal., 2001, 198: 1.
[26] Pande N K, Bell A T. J. Catal. 1986, 97: 137.
[27] Okuhara T, Misono M. Hyoumen. 1990, 28: 87.