http://www.chemistrymag.org/cji/2003/055041pc.htm |
May 1, 2003 Vol.5 No.5 P.41 Copyright |
(1Department of Chemistry, Liaoning Normal University, Dalian 116029, China; 2State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Science, Dalian 116023, China)
Abstract Because of the absence of
solution effect, gas-phase reaction of metal ion and organic molecule has become an
effective method to search for reaction mechanism and metal organic synthesis. With the
recently published technical literature as information source, this paper introduces the
gas-phase reaction of metal ion and organic molecule from two aspects, experimental method
and reaction mechanism. Focus is on the reaction mechanism.
Keyword Metal ion, Organic molecule, Gas-phase reaction, Reaction mechanism
(1辽宁师范大学化学系,辽宁 大连 116029; 2中科院大连化学物理研究所分子反应动力学国家重点实验室,辽宁 大连 116023)
2003年3月14日收稿
摘要
由于消除了溶剂效应的影响, 金属离子与有机分子的气相反应已经发展成为探索反应机理和金属有机合成的一个有效手段。本文以国内外近年来公开发表的科技文献为信息来源,结合我们的一些工作,从实验方法和反应机理两个方面介绍了金属离子与有机分子的气相反应,而着重介绍了反应机理。关键词 金属离子 有机分子 气相反应 机理
1 引言
近年来,由于消除了通常凝聚相中遇到的溶剂效应影响,气相反应已经发展成为探索金属有机化学反应机理和金属有机合成的一个有效手段。 金属离子与有机化合物分子的气相反应也成为金属有机化学的一个重要领域[1]。尤其是过渡金属离子与碳氢化合物的气相反应引起了人们极大的兴趣。在这类反应中,反应中间体与C-C键、C-H键的活化有关,这对于研究催化反应[2]的中间体具有重要意义。大家正努力认识C-H和C-C键活化的机理和一般的催化过程,了解含金属的离子和中性化合物的基本特性,复合物对金属中心和有机物残基反应性的影响,寻找高效化学离子化反应剂。金属催化反应不仅有基础研究价值, 还有应用价值。 过渡金属离子和烯烃的反应在很多均相和多相催化过程中起重要的作用,例如不饱和烃的氢化和烯烃的聚合。然而,目前这些反应的机理还不清楚。 一个对这些系统更深入了解的方法是在没有配体和溶剂的影响下研究化学反应。自从七十年代中期J. Allison等人在Fe+与i-C4H10气相反应中观察到失去烃分子的现象以后,人们对这类反应进行了大量研究,其中包括Ni+与小分子烷烃的反应,Fe+、Co+、Ni+与环状烃的反应,Fe+与烯烃的反应,Ni+与芳香族化合物的反应。不仅有实验研究,还有理论研究,例如张冬菊等用密度泛函方法(DFT)研究了Ni+和乙烯反应的机理[3]。本文结合我们的一些工作,对金属离子与有机分子的气相反应进行了综述。
2 实验方法
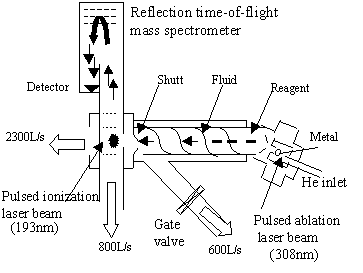
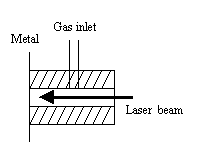
这种反应一般是在质谱仪内通过不同的离子源,将金属元素的单质或化合物电离成离子,再让离子与中性有机物分子反应,生成产物离子经分离检测,从而获得反应机理、化合物结构、键能等重要信息,同时,它还为金属有机物的合成提供了新的反应途径。一般来说,使用的离子源都是采用激光脱附技术来产生金属离子,然后进一步与有机分子反应,如图1所示。而R.J.Crutchley和A.B.P.Lever[4]使用电子轰击Fe(CO)5或Fe2(CO)9,来产生Fe+和Fe(CO)+,进一步与丁烷反应。L.F.Halle和P.B.Armentrout 等[5]使用的离子源是铼离子化灯丝。金属离子与有机分子的反应常常是在流管反应器或反应池[6,7]中进行的,这样可以保证反应的充分,如图2。研究这类反应目前还使用昂贵的离子回旋共振技术(ICR)[8]、傅立叶质谱技术(FT-MS)[6,7,9]、离子束装置[5]、激光烧蚀[10-13]、TOF-MS技术[11,13]和碰撞诱导解离方法(CID)[1,8]。
 |
 |
| 图1
激光烧蚀簇源和流管反应装置示意图[11] Fig.1 schematic diagram of the laser ablation |
图2 反应室示意图[15] Fig.2 Schematic diagram of reactor |
3 反应机理
关于金属离子与有机分子气相反应的机理,有很多种解释。通常认为金属离子与有机分子的气相反应是插入机理,分三步进行:(1),金属离子插入有机分子的键中;(2),发生异构化;(3),失去中性分子得到金属的络合物离子。具体反应的活性又和很多因素有关,例如金属离子的电子结构[12]、有机分子的烃链长短[9,10]、伸展程度[7]、分子结构、气压的大小[11]、环境温度[11]等。有的反应并不符合以上机理,例如,有的反应是金属离子先与有机分子形成络合物,然后异构化,再失去一个中性分子生成金属的络合物离子;或者金属离子与有机分子反应时,不失去中性分子,也不异构化,而是直接形成络合物离子,我们的实验就是这种情况。下面根据不同金属离子来介绍金属离子与有机分子的气相反应。
3.1 双原子金属离子
对于双原子金属离子与有机分子的气相反应,研究较多的是杂核双原子金属离子。一般来说,杂核双原子金属离子与有机分子反应的反应通道都是脱氢。1987年E.C.Tews和B.S.Freiser利用傅立叶变换质谱研究了气相中异核双原子过渡金属离子CuFe+和许多碳氢化合物的反应[9]。发现CuFe+与直链(C1―C6)和环状(C3―C6)烷烃不反应。1991年Lisa
M.Roth等在气相中异核双原子金属团簇离子MgFe+和与烃反应的研究中[10],也有类似发现,MgFe+不与小烷烃(C2―C4)反应,但和大烷烃(C5―C7)反应,形成MgFe+
―烯烃产物,反应通道是脱氢。在和烯烃反应时,E.C.Tews 和 B.S.Freiser观察到CuFe+能和带有支链C4单元的脂肪族烯烃反应[9],主要也是脱氢。而唯一能发生C-C键裂解甲烷消除反应的脂肪族烯烃是2,3,3-三甲基-1-丁烯。与CuFe+不同,在MgFe+团簇与直链烯烃(C2―C6)反应中,烯烃取代Mg形成Fe+
MgFe+也与环烯烃反应,例如,它使环己烯脱氢形成MgFe+―苯,
它进一步反应取代 Mg而形成苯―Fe+―环己烯。MgFe+还与乙醇反应,通过铁的取代形成Mg+―乙醇产物[10]。
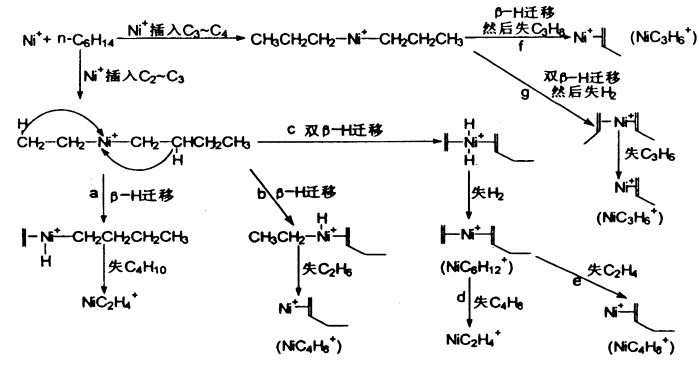
3.2 镍离子
国内张福义等对Ni+与一系列有机分子的气相反应作了大量研究,如醇类、酯类、酮类、烷烃等,总的来说是插入机理,分三步进行:(1),金属离子插入有机分子的C-C键或C-H键;(2),发生异构化,b-H或b-烷基迁移至Ni+上;(3),失去中性分子得到Ni+的络合物离子。在和醇反应时,Ni+还可以插入O―H键;和酮类反应时,Ni+有选择地插入O=C―C的C―C键中;和酯类的反应是Ni+与酯形成激发态络合物,然后b-H迁移至烃氧基的氧上形成Ni+的双配体化合物离子,再失去一个中性配体分子得到Ni+的单配体化合物离子(如上式)。
Zhang Dongju等用密度泛函理论研究了气相中Ni+(2D)与乙烷反应的机理[3]。提出了在Ni+和乙烷反应中生成产物CH4和H2的还原消除是典型的加合-消除反应。每一个反应都有两个基本步骤:C-C或C-H键活化形成插入物种,伴随异构化,形成类似产物的中间体。决定形成CH4和H2的消除反应步骤的速率是插入物种的异构化,而不是C-C或C-H键活化。发现形成H2的消除反应在热动力学上比CH4的更有利。D.B.Jacobson和B.S.Freiser发现Ni+对裂解内部键有很高的选择性,对插入末端C-C键也有很高的选择性。当有b-氢化物从第二个碳而不是从第一个碳上转移发生时,脱氢比脱烷烃更有利[8]。L.F.Halle等利用离子束装置研究了镍离子与几种烷烃的气相反应[5]。涉及C-C和C-H氧化加成到金属离子作为第一步的机理解释了这个研究中的所有产物。在与含杂原子的有机分子反应中,L.Rodriguez-Santiago等采用质谱和B3LYP密度泛函方法研究了气相中Ni+和氨基乙酸的反应[14],分析了[Ni-氨基乙酸]+的MIKE光谱,表明最重要的片段对应于水的失去,其他片段也有,但很少,即CO或CH2O2的脱去。并在B3LYP水平上研究了产生这些片段可能的机理,在所有这些机理中,最有说服力的是Ni+离子插入最稳定的h2-N,O结构的C-C键中。
3.3 镧系和锕系离子
金属离子与有机分子反应的研究,最初主要集中在d区过渡元素,自从Schilling
和Beauchamp[15]对镧系离子的研究揭示了不同的镧系离子的反应活性有很大差别(和凝聚态有机镧系化学相比[16])后,f区镧系和锕系元素才引起人们的注意。随后的研究证实了最初的猜想[15],不同的Ln+反应性反映了把基态离子激发到一个有两个非4f价电子的二价构型所需的能量,这个构型可以使Ln+插入C-H键中形成一个C-Ln+-H活化中间体。
John K. Gibson对此类反应研究的比较多。1998年John K. Gibson研究了镅离子Am+和烯烃的气相反应[12]。对于直链和环状C6烯烃,发现Am+相当有惰性,只有很少量的{Am+-苯}凝聚产物被检测到。与1,5-环辛二烯和环辛四烯的反应生成了脱氢复合物[Am-C8H8]+和[Am-C8H6]+,尽管和更轻的An+的相比少了许多。总体来说,Am+的C-H活化效率比在前面的An+要小,但更接近于相对不反应的镧系离子Tm+的C-H活化效率。这个结果说明,和Ln+的4f电子一样,Am+的5f电子没有完全参加C-H(或C-C)活化。很明显,Am+的[Rn]5f77s1占据轨道的基态必须被激发到[Rn]5f66d17s1态,比基态高245KJ/mol,这个激发态由两个有化学活性的6d/7s电子组成,能形成一个C-Am+-H脱氢中间体。同年John
K. Gibson 又和 Richard G. Haire 报道了气相中金属离子Cm+、 U+、Tb+与烯烃、乙腈和六氟丙烯反应[17]。重点放在和烯烃的反应上,尤其是脱氢,作为一个M+激活C-H键能力的标志(C-C激活的程度一般来说类似于C-H激活),与乙腈和所有烯烃(除了乙烯)反应,三种M+离子都导致了脱氢。对于不同反应物,差别是明显的,表1为Cm+,U+,Tb+与几种烯烃的脱氢碎片形成的络合物,总的脱氢效率的顺序是U+>Tb+>Cm+。这个顺序和电子激发能量的差异是一致的。这个能量能用来供给在M+也就是一个[Rg]fn-2d1s1构型上两个未成对的、非f价电子,使它插入一个C-H键。相对于U+和Tb+,
Cm+反应活性降低表明基态Cm+(8S[Rn]5f77s2)的闭壳7s2电子不能活化C-H键,激发到10D[Rn]5f76d17s2构型是先决条件。
Table 1 Abundance of the ML+ for the specified L from Propene, 1-Butene, and Isobutene[17]
Reactant |
Propene |
1-Butene |
Isobutene |
Metal Ion |
M(C3H4)+ |
M(C4H6)+ |
M(C4H6)+ |
Cm+ |
10 |
6.3 |
130 |
U+ |
500 |
130 |
360 |
Tb+ |
55 |
78 |
230 |
1999年John.K.Gibson用具有快速反应和检测的激光脱附技术进一步研究了锕系(M=U、Np、Pu和Am)和镧系(M=Tb和Tm)离子,M+与醇、硫醇、醚的气相反应[13]。和醇反应主要产物包括氢氧化物和醇盐、M(OH)1,2+、M(OR)1,2+、MO(OH)+和MO(OR)+,认为这些产物是来自于脱附的M+ 解离了RO-H和R-OH键。根据锕系和镧系离子不能使烷烃脱氢,醇的烃链脱氢的表现,就象从丙醇产生的HO-Pu+-C3H5O一样,表明了反应物离子与醇复合时的非插入机理。和OH从醇中提取出来生成金属氢氧化物相类比,从硫醇中SH提取出来产生了氢硫化合物,包括Am(SH)+和Np(SH)2+。和二甲基醚反应的初级产物是来自于C-O键裂解的甲醇盐。和甲基乙烯基醚反应,有更复杂的通道,大部分是稳定的有机分子消除。
3.4 3d过渡金属离子
3d过渡金属离子和烷烃反应主要是脱氢[6,18],和醇反应的通道比较多,有脱氢、脱水、还有脱小烷烃等[7]。而反应性又与不同的金属离子有关,不同的3d过渡金属离子呈现不同的反应性和选择性。在我们的实验中,Cu+与丙酮的气相反应只形成了络合产物,并未见到其它产物。1998年Asgeir
Bjarnason和D.P.Ridge使用傅立叶变换质谱描绘了M+(M=Sc,Ti,V)与甲苯在气相中的反应[6]。在Sc+和Ti+与甲苯反应生成MC7H6+的反应中脱氢是主要的反应通道。但V+不能使甲苯脱氢,只能形成加合产物。为了进一步描绘这些反应,也研究了C6H5CD3和甲苯-a-13C,没想到的是观察到了在他们和Sc+的反应中与和Ti+反应相比有不同的行为。在和C6H5CD3的反应中,Sc+主要消除HD(90%)和少量H2(8%)、D2(2%),然而Ti+呈现很少的选择性,消除HD(50%)、H2(20%)、D2(30%)三种不同产物离子。见表2。
Table 2 Products Ion Distribution from the Primary Reactions of Sc+, Ti+, and V+ with Toluene and Toluene-d3[6]
|
|||||
C6H5CH3 C6H5CD3 |
|||||
Metal ion |
-H2 |
M(toluene)+ |
-H2 |
-HD |
-D2 |
Sc+ |
100 |
0 |
8 |
90 |
2 |
Ti+ |
70 |
30 |
20 |
50 |
30 |
V+ |
0 |
100 |
|||
Y.H.Pan等报道了环己烷和Co+、Co2+、Co3+、Co4(CO)n+(n=0-12)、Ir2+、Ir3+双分子气相反应的速率常数和产物分配[18]。多数情况下是脱氢,主导产物是C6H6和金属团簇相连。1986年Sunkwei
Huang等利用傅立叶变换质谱研究了由多光子电离产生的气相过渡金属离子Fe+、Cr+和Mo+与一系列脂肪族醇的反应[7]。这些金属离子进行三类键插入反应:C-O插入(脱水)、C-H或O-H插入(脱氢)和C-C插入。总的反应性与金属离子的电子结构、醇的脂肪链长度和伸展程度有关。对于直链含有三个或更多碳的醇,反应性顺序是Fe+>Mo+>Cr+。见图3。对于更短的醇,Mo+反应性最强,而Cr+与甲醇和乙醇不反应。1982年L.F.Halle等利用离子束装置,研究了铁离子与几种烷烃的气相反应[5]。涉及C-C和C-H氧化加成到金属离子作为第一步的机理解释了这个研究中的所有产物。
1989年Ben.S.Freiser利有傅立叶变换质谱研究了气相中Fe+-苯和它与小烷烃反应性的单分子化学[1]。Fe+-苯的碰撞诱导解离只有苯的消失。相比较而言,Fe+-苯的光解不仅产生了苯和Fe+的解离,还有C2H2和C2H4的竞争消除。所有产物离子都可以用基于第一步Fe+插入C-C或C-H键中,然后,甲基或H从Fe+迁移到苯配位,或者其中之一,苯迁移插入一个金属-C或金属-H键中。
3.5 其他金属离子
David B. Pedersen等利用激光烧蚀金属团簇源和激光电离/飞行时间质谱探测,耦合大孔流管反应器,测量了Wn和环丙烷反应的绝对二阶速率常数[11]。
W团簇被He载气中环丙烷耗尽的速率常数显示出在0.5到2.0Torr之间不存在He气压力依赖关系,但是在277
和 351k
之间随着温度的升高而降低。这说明金属离子与有机分子的反应性与载气压力和环境温度也有一定的关系。
Peter A.Willis等利用交叉分子束技术,研究了过渡金属离子Zr+和Nb+与乙烯的反应[19]。使用电子轰击和157nm光电离质谱在5到23kcal/mol的碰撞能量下测量了脱去H2后MC2H2产物的角分布和速度分布。在碰撞能小于6kcal/mol时,发现来自于ZrC2H4衰减的Zr的非反应色散可以忽略,然而这个通道对于Nb来说是不能的。MC2H4复合物衰减回M+C2H4和插入C-H键形成HMC2H3之间竞争的RRKM模型表明对于Zr的情况存在一个M和C2H4结合的绝热势垒,这个过程的过渡态比Nb+C2H4的相似过程更加困难。Zr和C2H4结合的势垒归因于Zr的排斥的s2基态构型,然而对于Nb,s1基态构型导致结合没有势垒。在低碰撞能下ZrC2H4没有衰减回Zr+C2H4的事实表明插入C-H键形成HZrC2H3的势垒低于Zr+C2H4结合的势垒,很可能是最初没有形成ZrC2H4而直接插入C-H键起了重要作用。
4 小结
金属离子和有机分子在液相中的反应很早以前就开始了,由于溶剂的影响,使人们对该类反应的机理并不太清楚。而气相反应正好可以消除溶剂的影响,为金属离子和有机分子反应机理的研究打开了道路。人们从不同角度探索了金属离子和有机分子的气相反应,有的从热力学,有的从动力学,还有的从电子结构等角度分析解释了反应所得的产物。尽管大家对于金属离子和有机分子的气相反应给与了极大关注,并做了很多研究,但目前并没有一个公认的反应机理可以很好地解释所有金属离子和有机分子的气相反应,还有很多问题有待解决。插入机理并不能解释所有反应,插入时也有一定的选择性,金属离子的选择性如何?键的选择性又怎样?而电子结构又怎样影响插入反应和选择性?这些问题的解决有赖于进一步探索相应的基元反应,并将为催化过程和有机合成提供坚实的基础。
REFERENCES
[1] Huang Y, Freiser B S. J Am Chem Soc, 1989, 111: 2387.
[2] Freiser B S. J Mass Spectrom, 1996, 31: 703.
[3] Zhang D J et al. Chinese J Chem, 2002, 20: 220.
[4] Crutchley R J et al. J Am Chem Soc, 1980, 102: 7129.
[5] Halle L F et al. Organometallics, 1982, 1: 963.
[6] Bjarnason A, Ridge D P. Organometallics, 1998, 17: 1889.
[7] Huang S et al. Organometallics, 1986, 5: 1857.
[8] Jacobson D B, Freiser B S. J Am Chem Soc., 1983, 105: 736-742.
[9] Tews E C, Freiser B S. J Am Chem Soc., 1987, 109: 15.
[10] Lisa M R et al. J Am Chem Soc, 1991, 113 (9): 3274.
[11] David B P et al. J Chem Phys, 1998, 109 (2): 551
[12] John K G. Organometallics, 1998, 17: 2583.
[13] John K G. J Mass Spectrom, 1999, 34: 1166.
[14] John K G, Richard G H. J Phys Chem A, 1998, 102: 10746.
[15] L RS et al. J Phys Chem A., 2001, 105: 5340.
[16] Liu Hong, Zhang Fuyi et al. Chinese Journal of Analytical Chemistry, 1998, 26 (3):
254.
[17] Schumann H, Genthe W. In Handbook on the Physics and Chemistry of Rare Earths, 1984,
7: 445.
[18] Peter A W et al. J Phys Chem A, 1999, 103: 3706.
[19] Pan Y H et al. J Am. Chem. Soc., 1991, 113 (7): 2406.