http://www.chemistrymag.org/cji/2003/057054pe.htm |
Jul. 1, 2003 Vol.5 No.7 P.54 Copyright |
Peng Changjun, Li Jiankang, Liu Honglai, Hu Ying
(Department of Chemistry, East China University of Science and Technology,
Shanghai, 200237, China)
Abstract Monte Carlo simulations for
the adsorption of block copolymers in nonselective solvent at solid-liquid interface have
been performed on a simple lattice model. In simulations, block copolymer molecules are
modeled as self-avoiding linear chains composed of m segments of A and n
segments of B and the segment A is attractive, while the segment B is non-attractive to
the surface. Block copolymer molecules are![]() ,
,![]() and
and![]() ,
respectively. The aim of this work is to provide a systematic comparison between diblock
and triblock copolymer to study the influence of the sequence of copolymer to adsorption
behavior including the size distribution of loops, tails and trains configurations, the
density profiles, the surface coverage, adsorbed amount and adsorption layer thickness.
The results show that the size distribution of train and tail configuration for
,
respectively. The aim of this work is to provide a systematic comparison between diblock
and triblock copolymer to study the influence of the sequence of copolymer to adsorption
behavior including the size distribution of loops, tails and trains configurations, the
density profiles, the surface coverage, adsorbed amount and adsorption layer thickness.
The results show that the size distribution of train and tail configuration for![]() ,
,![]() and
and![]() all present two peaks. The peak intensity and
position is determined by the sequence of copolymer and the reduced adsorption energy.
all present two peaks. The peak intensity and
position is determined by the sequence of copolymer and the reduced adsorption energy. ![]() can easily form bigger loop configuration. The
surface coverage and adsorbed amount are also affected by the sequence of copolymer.
Copolymer
can easily form bigger loop configuration. The
surface coverage and adsorbed amount are also affected by the sequence of copolymer.
Copolymer ![]() has the highest adsorbed amount,
while copolymer
has the highest adsorbed amount,
while copolymer![]() always gives the smallest
surface coverage and adsorbed amount as well as adsorbed layer thickness.
always gives the smallest
surface coverage and adsorbed amount as well as adsorbed layer thickness.
Keywords Block copolymers, Surface adsorption, Monte Carlo simulation
The behavior of polymeric materials near a solid surface is very different from that in the bulk because of the existences of strong entropic constraints placed upon the polymer chain conformations near a bounding impenetrable surface. Many technological processes such as the stabilization of various commercial dispersions, surface treatment of materials and lubrication need understanding of the microscopic information around an interface, especially, the microstructure of adsorption layers in which polymers interact with the materials. Block copolymers composed of two or more kinds of segments with different physical and chemical properties usually exhibit amphiphilic behavior. This makes them a good model for the study of surfactants, especially nonionic and amphiphilic surfactants. A series of computer simulations of adsorption behavior have been reported in the literatures for diblock copolymers[1-9] and for triblock copolymers[10-13], Much information about microstructure of adsorbed layers including different adsorbed configurations and their distribution can be provided naturally and intuitively with the aid of computer simulation. Recently, a review on the simulation studies of polymer blends at interface has been given by Muller and Schmid[7]. In the following we briefly mention some examples about simulation studies of adsorption behavior for triblock copolymer.
Balazs and Lewandowski[10] have developed computer simulations to model the adsorption of
From the above brief reviews, it is obvious that there are some progresses on the study of adsorption of triblock copolymers at interface, but the adsorption of triblock copolymer with the middle block sticky has rarely been studied and the size distribution of different configurations is not addressed adequately. In this work, we will focus our attention on the microstructure and adsorption layer information of block copolymer at solid-liquid interface by simulation. The block copolymers are either only one sticky end or two sticky ends or middle-attractive. The density profiles and the size distributions of tail, loop and train configurations are studied. The adsorption information such as the surface coverage and adsorption amount as well as adsorption layer thickness are also presented. The aim is to compare the influence on adsorption behavior at solid-liquid interface by the different sequence of copolymer. 2. PHYSICAL MODEL AND SIMULATION METHOD
Three types of block copolymers,
The number density of chains, Fc, the segment density or concentration at layer Z,
where M, NZ , Na and r are the number of chains introduced into the simulation box, the number of segments in the Z-th layer, the number of adsorbed chains on a surface and the chain length, respectively,
The surface coverage,q , is defined as the segment density or concentration at layer Z=1 or Z=LZ, i.e. q = f1 or q = fLz. qA and qB are the corresponding segment densities of A and B, respectively.
The bulk concentration, f0, i.e. the segment density of the region where the concentration of segments is constant among with lattice layers, can be determined by averaging the most intermediate 21 layers because adsorption layers typically extend up to approximately 10 layers away from the surface. The intermediate 21 layers are between 15 and 35 for 50 lattice layers. 3. RESULTS AND DISCUSSIONS
3.1 Size distributions of various configurations of adsorbed block copolymers
The chains adsorbed usually present in various configurations, which further determine the other adsorption properties. We thus will first present the chain configurations. When the numbers of attractive and non-attractive segments are all fixed, the size distributions of various configurations would have a large difference as expected if the position of attractive blocks in a copolymer chain is different. Size distributions of different configurations for diblock A10B20, triblocks A5B20A5 and B10A10B10 for
 |
 |
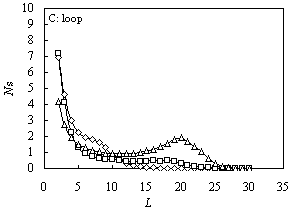 |
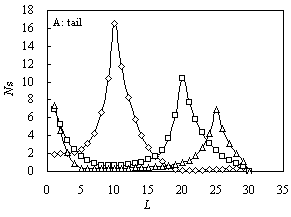
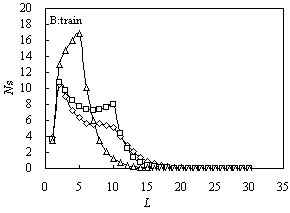
Fig. 1 The size distribution
of various configurations for A10B20 (square), A5B20A5
(triangle) and B10A10B10 (diamond) for ![]() and
and![]() .
A: tail; B: train; C: loop
.
A: tail; B: train; C: loop
The size distributions of
tails for the three samples have two peaks as shown in Figure 1(A). For B10A10B10,
the first peak appears at L = 10, which just equals to the length of non-attractive
block B; the other small one is at L = 29 corresponding to the case when only one
segment is adsorbed, where L is the length of configuration. The second peak of B10A10B10
would become larger for smaller energy. However, for A10B20 and A5B20A5,
the first peak all appears at L = 1, and the second peak of A10B20
is at L =20 while A5B20A5 is at L = 25.
The size distributions of trains for the three samples have a platform,
from L =2 to 5 for A5B20A5, from L = 2 to
10 for A10B20 and B10A10B10, as
shown in Figure 1(B). The platform of A10B20 is higher than that of
B10A10B10. With the increasing of L, the
distribution curve decreases faster for the former than for the latter. From Figure 1(B),
the size distributions of trains are determined by attractive block. The first peak always
appears at L=2, the second peak appears at the length of a segment of attractive
block; but whether the second peak of train configuration appear or not is determined by
energy between segment and surface. If the length is shorter, the emergence of the second
peak of trains only needs a small energy.
The size distributions of loops are shown in Figure 1(C). For A10B20
and B10A10B10, just one peak appears at L =
2, then it will decrease monotonically. For A5B20A5,
there are two peaks at L = 2 and L = 20, indicating more chance to form
bigger loop configuration when both the terminals are attractive.
3.2 Density profiles
The segment-density profiles of three systems with respect to the distance from the
surface,![]() , for
, for![]() and
and![]() are depicted in Figure 2. Profiles include that of segments A and B. From the figure we
can see that segments A form a thin adsorbed layer directly adjacent to the surface by
manifesting a maximum density in the first layer. Obviously, this is because of the
attractive interaction with the surface enhancing a preferential adsorption. With the
increase of distance from the surface, density of segment A decreases very quickly and
then increases a little to approach the bulk value. In the first layer, the largest
density is achieved by A10B20, then by B10A10B10,
and the smallest by A5B20A5. With the increasing of
distance from the surface, the position of density profiles between A10B20
and A5B20A5 would be exchanged, but B10A10B10
is still in the middle.
are depicted in Figure 2. Profiles include that of segments A and B. From the figure we
can see that segments A form a thin adsorbed layer directly adjacent to the surface by
manifesting a maximum density in the first layer. Obviously, this is because of the
attractive interaction with the surface enhancing a preferential adsorption. With the
increase of distance from the surface, density of segment A decreases very quickly and
then increases a little to approach the bulk value. In the first layer, the largest
density is achieved by A10B20, then by B10A10B10,
and the smallest by A5B20A5. With the increasing of
distance from the surface, the position of density profiles between A10B20
and A5B20A5 would be exchanged, but B10A10B10
is still in the middle.
Segments B exhibit a low density in the first layer because they are
non-attractive. From this figure, we can see that segment B, although non-attractive, can
still be adsorbed onto the surface with small adsorption amount due to chain connectivity.
The density of segment B then increases with the increment of distance from the surface
and reaches a maximum value at several layers away from the surface due to the limitation
of chain connectivity. The exact position of the maximum is determined mainly by the
position of attractive block located and the length of B block. It is at the 3rd layer for
B10A10B10 and A5B20A5,
and the 4rd layer for A10B20. After the maximum is reached, the
density decreases monotonically to the bulk value.

Fig. 2 The density profiles of segment A (open symbol) and segment B
(solid symbol) for ![]() and
and![]() . Diamond: A10B20; square:
A5B20A5; triangle: B10A10B10
. Diamond: A10B20; square:
A5B20A5; triangle: B10A10B10
3.3 Surface Coverage and Adsorption Isotherms
Figure 3 illustrates surface coverage under other different bulk concentration for A10B20,
A5B20A5 and B10A10B10
for ![]() . From
simulation, we found that the surface coverage of segment A for three samples are all
increased monotonously with the bulk concentration, when the reduced adsorption energy
between segment A and surface is 1.2. However, the relative position is rather different
from each other. The upper one is for A10B20, the middle for B10A10B10
and the lower for A5B20A5, respectively. If energy is
smaller, the surface coverage of A10B20 is usually greater than that
of B10A10B10 and A5B20A5.
The surface coverage for the latter two is nearly the same.
. From
simulation, we found that the surface coverage of segment A for three samples are all
increased monotonously with the bulk concentration, when the reduced adsorption energy
between segment A and surface is 1.2. However, the relative position is rather different
from each other. The upper one is for A10B20, the middle for B10A10B10
and the lower for A5B20A5, respectively. If energy is
smaller, the surface coverage of A10B20 is usually greater than that
of B10A10B10 and A5B20A5.
The surface coverage for the latter two is nearly the same.
The value of surface coverage depends also on the relative number of
attractive and non-attractive segments. We have also studied two other special series, one
is A18B2, A9B2A9 and B1A18B1,
and another is A2B20, A1B20A1 and B10A2B10.
The former has a rather shorter non-attractive block and the latter attractive block. For
series of A18B2, A9B2A9 and B1A18B1,
the difference of surface coverage between each other is not so large and can be
neglected. It means that when non-attractive segment block is much shorter than attractive
one, the influence of sequence of copolymer on surface coverage is not great. For series
of A2B20, A1B20A1 and B10A2B10,
the surface coverage due to segment A is ordered in value from B10A2B10,
A1B20A1 to A2B20 for![]() . If energy is further reached to 1.2, this order
would be A1B20A1, B10A2B10
and A2B20.
. If energy is further reached to 1.2, this order
would be A1B20A1, B10A2B10
and A2B20.

Fig. 3 The surface coverage duo to segments A for A10B20 (diamond),
A5B20A5 (square) and B10A10B10
(triangle) for ![]() and
and![]() .
.
Adsorption isotherms for diblock A10B20,
triblocks A5B20A5 and B10A10B10
are plotted in Figure 4. The adsorption amount increases with the bulk concentration for
all systems. It is found that when![]() , the
adsorbed amount is greater for the diblock copolymer A10B20 than for
the triblock copolymer A5B20A5 and B10A10B10.
Especially, the adsorbed amount of B10A10B10 is greater
than that of A5B20A5 if
, the
adsorbed amount is greater for the diblock copolymer A10B20 than for
the triblock copolymer A5B20A5 and B10A10B10.
Especially, the adsorbed amount of B10A10B10 is greater
than that of A5B20A5 if![]() , and the former is even lower than the latter as
, and the former is even lower than the latter as ![]() further increases. The adsorbed amount is
ordered in value from B10A10B10, A5B20A5
to A10B20 when
further increases. The adsorbed amount is
ordered in value from B10A10B10, A5B20A5
to A10B20 when![]() .
However, the difference between A10B20 and A5B20A5
is very small.
.
However, the difference between A10B20 and A5B20A5
is very small.


Fig. 4 The adsorption isotherms for A10B20 (diamond), A5B20A5
(square) and B10A10B10 (triangle) for![]() and
and![]() .
.
Fig. 5 The adsorption isotherms for A2B20 (diamond), A1B20A1
(square) and B10A2B10 (triangle) for![]() and
and![]() .
.
For series of A18B2,
A9B2A9 and B1A18B1, the
adsorbed amount is nearly the same whatever energy is used. This implies that the
influence of the sequence of copolymer on adsorbed amount can be neglected. For series of
A2B20, A1B20A1 and B10A2B10,
A2B20 and A1B20A1 give nearly the
same adsorbed amount which is slightly greater than B10A2B10
does when![]() . If
. If![]() , the biggest adsorbed amount is achieved by A2B20,
followed by A1B20A1 and B10A2B10
as shown in Figure5. Therefore, the order of adsorbed amount is
, the biggest adsorbed amount is achieved by A2B20,
followed by A1B20A1 and B10A2B10
as shown in Figure5. Therefore, the order of adsorbed amount is![]() ,
, ![]() and
and![]() . This conclusion is
inconsistent with that of Haliloglu et al13, who reckoned that, at low
. This conclusion is
inconsistent with that of Haliloglu et al13, who reckoned that, at low![]() , the adsorbed amount is greater for
, the adsorbed amount is greater for![]() than for
than for![]() because the surface has a higher chance to meet any one of the two sticky
ends belongs to a triblock than only one end of the diblock copolymer chain. However, our
simulation results show explicitly that the adsorbed amount is commonly greater for
because the surface has a higher chance to meet any one of the two sticky
ends belongs to a triblock than only one end of the diblock copolymer chain. However, our
simulation results show explicitly that the adsorbed amount is commonly greater for ![]() than for
than for![]() . This can be explained as follows:
. This can be explained as follows:![]() has more chance to form bigger loop configuration when both the two ends
are attractive; loop configuration will counteract more attractive segments approaching
surface. In other words, a series of loop configurations can lead to screening effects
near the interface, and the adsorbed amount then decreases.
has more chance to form bigger loop configuration when both the two ends
are attractive; loop configuration will counteract more attractive segments approaching
surface. In other words, a series of loop configurations can lead to screening effects
near the interface, and the adsorbed amount then decreases.
3.4 Adsorption layer thickness
The results simulated show that the thickness of tails always greater than that of loop,
being independent of the sequence of copolymer. It implies that tails play a more
important role in determining the thickness of adsorption layer. The number and size of
tails together determine the adsorption layer thickness. The adsorption layer thickness
versus bulk concentration is depicted in Figure 6 for different energies for diblock A10B20, triblocks A5B20A5
and B10A10B10. Thickness decreases monotonously for three
systems, with the increasing of bulk concentration when energy is 0.4. The thickness is
the largest for A5B20A5 and the smallest for B10A10B10.
If energy is 1.2, The thickness first increases quickly and then slowly. In this case, the
largest thickness is for A10B20 because A5B20A5
forms bigger loop.

Fig. 6 The adsorption layer thickness for A10B20 (diamond),
A5B20A5 (square) and B10A10B10
(triangle) when ![]() (dash line) and
(dash line) and ![]() (solid line)
(solid line)
For series of A2B20, A1B20A1 and B10A2B10, the thickness of A2B20 and A1B20A1 is nearly the same and all greater than that of B10A2B10 if energy is 0.4. When the energy becomes larger, the thickness is ordered in value from B10A2B10, A1B20A1 to A2B20. For series of A18B2, A9B2A9 and B1A18B1, whatever energy is used, their thickness has the sequence of order always as B1A18B1, A18B2 and A9B2A9.
4. CONCLUSIONSExtensive Monte Carlo simulations on a cubic lattice have been performed to provide a systematic comparison between diblock and triblock copolymer to study the influence of the sequence of copolymer to adsorption behavior. The results simulated show that the position of attractive block in a copolymer chain affects the adsorption behavior.
It is found that the size distribution of train and tail for
[1] Zhan Y, Mattice W L, Napper D H. J. Chem. Phys., 1993, 98: 7502.
[2] Clancy T C, Webber S E. Macromolecules, 1997, 30:1340.
[3] Wang Y, Li Y, Mattice W L. J. Chem. Phys., 1993,98: 9881.
[4] Wang Y, Li Y, Mattice W L. J. Chem. Phys., 1993,99: 4068.
[5] Wang Y, Teraoka I. Macromolecules, 2000, 33: 3478.
[6] Kim S H, Jo W H. J. Chem. Phys., 1999,110:12193.
[7] Muller M, Schmid F. Annu. Rev. Comput. Phys., 1999, 6: 59.
[8] Geisinger T, Muller M, Binder K. J. Chem. Phys., 1999,111: 5241.
[9] Wang Q, Yan Q, Nealey P F, et al. J. Chem. Phys., 2000, 112: 450.
[10] Balazs A C, Lewandowshi S. Macromolecules, 1990, 23: 839.
[11] Misra S, Mattice W L. Macromolecules, 1994, 27: 2058.
[12] Nguyen-Misra M, Misra S, Mattice W L. Macromolecules, 1996, 29:1407.
[13] Haliloglu T, Stevenson D C, Mattice W L. J. Chem. Phys., 1997, 106: 3365.
[14] Jiang J W, Liu H L, Hu Y. Macrom. Theory Simul., 1998, 7: 113.
[15] Chen T, Liu H L, Hu Y. J. Chem. Phys., 2001, 114: 5937.