http://www.chemistrymag.org/cji/2003/058068re.htm |
Aug. 1, 2003 Vol.5 No.8 P.68 Copyright |
Recent progress in two-dimensional organized molecular assemblies
Du Xuezhong, Liang Yingqiu(Laboratory of Mesoscopic Materials Science, College of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China)
Received Jun. 24, 2003; Supported by the Natural Science Foundation of China (No. 20073019, 29473100, 29873022, and 20273029).
Abstract Our recent advance in molecular
structures and properties of two-dimensional functional and biological organized molecular
assemblies is reviewed. A new mechanism of bilayer membrane formation from simple
single-chain amphiphiles without rigid segment was proposed and demonstrated: the
interaction between hydrophilic headgroups through hydrogen bond, ionic bond, and metal
complex induces a parallel packing of the corresponding hydrophobic chains. The new
mechanism was suitable to the two-dimensional organized molecular assemblies not only in
aqueous solutions and but also in organic phases, including the monolayers of
bolaamphiphile, metal complex, conjugated polymer, and coordination polymer and organogels
(bilayer membranes) through ingenious molecular design. It is shown that the organized
molecular assemblies in various media can be achieved. Surface-enhanced Raman scattering
(SERS) and in-situ infrared reflection absorption spectroscopy (IRRAS) techniques were
successfully introduced to study the molecular recognition of nucleolipid monolayers to
complementary bases in the subphase. The exact sites for the hydrogen-bonding interaction
between base pairs (A=T and G≡C) were presented, and
the photochemical reactions of relevant bases at room temperature were achieved.
Keywords Molecular assemblies; Bilayer membranes; Organogels; Monolayer membranes;
Langmuir- Blodgett films.
1. INTRODUCTION
Cell membrane, with the interplay of molecular self-organization and molecular recognition
to assemble systems, is one of the most perfect examples for functional supramolecular
systems[1]. The functions of the supramolecular systems are based on the
performance of their organization rather than on properties of single molecules. Membrane
mimic chemistry is located at the interface between life science and material science. It
applies the principles of self-organization, regulation, communication, and cooperation
and has advanced to a promising area of applied science undermining the borderline between
scientific disciplines and offering new routes for the design of materials where the
organization precedes the function[2-4]. Two-dimensional (2D) self-organized
molecular assemblies provide the microenvironment and molecular arrangement similar to
cell membranes and possess ordered structures at the molecular level to be main materials
in constructing molecular devices. It is of significant importance to characterize the
structures and elucidate the structure-function relationships of organized molecular
assemblies to understand the complicated processes occurring in them. This review reports
our recent research progress in 2D self-organized molecular assemblies since 1996. The
research advance earlier than this review was presented elsewhere[5].
2. FUNCTIONAL MOLECULAR ASSEMBLIES
2.1. Bilayer Membranes from Simple Single-chain Amphiphiles
2.1.1. Vesicle formation in aqueous solutions
Synthetic bilayer membrane formed from didodecyldimethylammonium bromide surfactants in
aqueous solution was first reported by Kunitake et al. in 1977[9]. A new
research field of synthetic bilayer membranes has been explored since then. It is commonly
considered that rigid segments (e.g., biphenyl, azobenzene, diphenylazomethine units) are
essential for single-chain amphiphiles to form stable bilayer membranes in dilute aqueous solutions[3,7-12], except for very few cases
such as hyperextended alkyl chains and perfluorinated chains. We recently reported stable
bilayer membranes formed from a series of simple amphiphiles with monoalkyl chains in
common length and without rigid segment in aqueous solutions[13,14], which has
explored a new research branch for the design and construction of synthetic bilayer
membranes.
The series of monoalkylethylenediamine amphiphiles 1 (n = 8, 12,
14, 16, and 18) could self-assemble into stable bilayer membranes in pure water and dilute
acidic solutions. The vesicular morphologies of the dispersions in dilute inorganic acid
solutions (HCl, HBr, and HClO4) were observed by transmission electron
microscope (TEM), and the size and density of the vesicles increased with extending chain
length[14]. The restricted molecular motion in synthetic bilayer membranes was
readily reflected by the line-broadening phenomenon in 1H NMR spectra. The
tighter the packing of membrane-forming molecules, the broader the line width is. It was
demonstrated that the hydrophilic headgroups and hydrophobic chains of amphiphiles 1
stacked closely in both pure water and acidic solutions with this method[13,14].
The gel-liquid crystalline phase transition is one of basic physicochemical properties of
bilayer membranes[3]. The phase transition temperature (Tc) and
enthalpy change of the amphiphiles 1 in pure water and acidic solutions (HCl, HBr,
and HClO4) exhibited a fairly good linearity with chain length, indicating that
the series of amphiphiles 1 formed bilayer structures bearing a similar packing
mode either in pure water or in acidic dispersion[14]. Moreover, Tc
decreased with increasing degree of protonation, in the order of unprotonated, partially
protonated, and perprotonated headgroups, and with increasing anion size (Cl- ,
Br- , and ClO4- )[13,14]. The
bilayer thickness of all samples, obtained from small-angle X-ray diffraction (XRD), also
displayed a good linearity with chain length. Comparing the bilayer thickness with the
evaluated molecular length by CPK model, the amphiphiles took the vertical tail-to-tail
(H-like) bilayer-packing mode in pure water and the tilted tail-to-tail (J-like)
bilayer-packing mode in acidic solutions[13,14]. Moreover, the protonated
headgroups and large-size anions gave rise to a tilt in chain orientation and a decrease
in Tc because the vertical chain-packing mode had a stronger interchain
interaction than the tilted chain-packing mode[3]. The XRD results obtained
were in good agreement with the differential scanning calorimetric (DSC) results. It was
shown that the formation of bilayer membranes from the simple single-chain amphiphiles 1
was owing to the parallel orientation arrangement of the hydrophobic chains induced by the
interaction (intermolecular hydrogen bonds and ionic bonds) between the hydrophilic
headgroups[13,14] (see Fig. 1).

Fig. 1 Structure-property relationship of
self-organized bilayer membranes formed from 1 in different aqueous solutions.
It is well known that
metal ions are readily coordinated with ethylenediamine derivatives. When amphiphiles 1
were dispersed in Cu2+-containing solutions. TEM observations (vesicular
morphologies) and NMR spectra (broadened line width) indicated that 1
self-organized into stable bilayer membranes in the Cu2+-containing solutions[15-18],
as in the cases of 1 in both pure water and acidic solutions[13,14].
However, these structures were quite different from bilayers formed in pure water or in
acidic solutions. The dispersion of n = 8 amphiphilic aggregates was clear blue in Cu(NO3)2
solution, and the others (n > 8) gave purple emulsion[15,16]. From the d-d
and s-s* transition
energies of electronic reflectance spectra of the corresponding cast films, it was deduced
that when n = 8, the Cu2+-coordinated headgroups adopted a trans-configuration
twisted tetrahedral CuN4 coordination with a horizontal orientation, and the
two tails in each complex took an approximately diagonal position, while for n > 8, the
complexed headgroups adopted cis-configuration square coplanar CuN4
coordination with a vertical orientation, and the two tails in each complex were located
at the same side of the plane and parallel to each other[15,16]. It is clear
that the headgroup structure has a great influence on the packing fashion of the
hydrocarbon tails in the organized aggregates. The bilayer thickness of the coordinated
films obtained from XRD patterns showed a good linearity with chain length for n > 8,
while n = 8, the bilayer thickness value was far above the straight line[15,16].
The results reflected the structural discrepancy between the cast bilayer membranes formed
by n = 8 and n > 8 amphiphiles. It was shown that the coordinated bilayer membrane (n =
8) took a tilted tail-to-tail packing mode, while in the case of n > 8 the molecules
probably assumed an interdigitated one[15,16]. The two types of chain packing
modes were further supported by the DSC results. The Tc for the n > 8
molecules was larger than 84 ºC, and exhibited a good linearity with chain length, and the Tc
for n = 8 molecules (about 40 ºC) was far below the straight line, which provided another evidence
for the tilted tail-to-tail chain packing mode[15,16]. It is obvious that the
molecular structure and phase transition property of the coordinated bilayer membranes
strongly depend on chain length of the amphiphiles.
In addition, it was found that the type of counter anion was of vital
importance to the determination of the self-organized structures. When amphiphiles 1
(n > 8) were dispersed in aqueous CuX2 (X = Cl- , Br-
, NO3- , and ClO4- )
solutions, the dispersions gave blue emulsion in the cases of Cl- and Br-
ions and purple emulsion in the cases of NO3- and ClO4-
ions[17,18]. On the basis of the d-d and s-s* transition energies of electronic reflectance spectra, it was
deduced that when X = Cl- and Br- ions, the Cu2+-coordinated
headgroups adopted a trans-configuration twisted tetrahedral CuN4 coordination,
and the two tails in per complex took an approximately diagonal position, while for X = NO3-
and ClO4- ions, a cis-configuration square coplanar
CuN4 coordination was adopted with the two tails located at the same side of
the plane and parallel to each other[17,18]. The bilayer thickness and Tc
of each system showed a good linearity with chain length, which indicated a similar chain
packing in the membrane structure of each system[17,18]. It has been shown that
in many cases the extensively tilted tail-to-tail type bilayer assemblies possess
relatively tilted chain packing and exhibited relatively low Tc, whereas the
interdigitated type bilayer membranes have tighter chain packing and display higher Tc
values than the tail-to-tail ones[3,19]. By combining the XRD and DSC results,
in the cases of X = Cl- and Br- ions, the Tc was lower
than 65 ºC, and the amphiphiles adopted a tail-to-tail chain packing mode
with the tilt angles 58.2° (Cl- ) and 61.6° (Br- ); while in the
cases of X = NO3- and ClO4- ions,
the Tc was higher than 60 ºC, and the amphiphiles took a partially interdigitated chain packing
mode with the tilt angles 33.4° (NO3- ) and 40.2° (ClO4-
)[17,18]. The schematic representation of bilayer formation from
amphiphiles 1 in aqueous CuX2 solutions is shown in Fig. 2. It is
clearly seen that the coordination of the headgroups with Cu2+ ions was of
significant importance to the determination of the membrane structures and properties.
It is obvious that the monoalkyl ethylenediamine amphiphiles form
stable bilayer membranes in pure water, acidic water, and metal-containing solutions
through the interactions of hydrogen bond, ionic bond, and metal complex between the
hydrophilic headgroups to induce an almost parallel arrangement of the corresponding
hydrophobic alkyl chains to achieve the balance of the hydrophilic and hydrophobic
interactions.
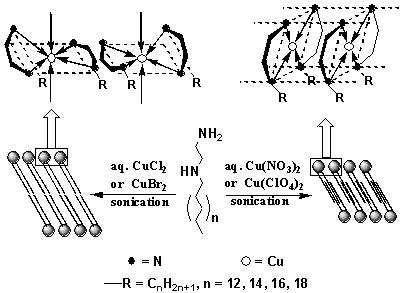
Fig. 2 Schematic representation of bilayer membranes
formed from 1 in aqueous solutions containing CuX2 (X = Cl- ,
Br- , NO3- , and ClO4-
).
Interestingly, we recently
found that some of common micelle-forming surfactants could self-organize into vesicles
induced by metal ions in dilute aqueous solutions. Sodium hexadecylimino diacetate 2
self-assembled into vesicles induced by Cu2+, Co2+, and Ni2+
ions in aqueous solutions[20]. Vesicular morphologies were observed with TEM
for the three dispersions. A combination of small-angle XRD and DSC results implied that
the hydrocarbon chains were packed in an interdigitated mode[20]. It was
deduced that 2 coordinated with Cu2+, Co2+, and Ni2+
ions to form double-tailed surfactants with a coordinated headgroup[20]. In the
cases of Cu2+ and Co2+ ions, the coordinated headgroups led to an
octahedral coordination with the two tails in the complexes located at the diagonal
positions of a parallelogram, while in the case of Ni2+ ion, a parallelogram
coordination model was adopted with the two tails in the complexes also located at the
diagonal positions[20], which were schematically shown in Fig. 3.
For the very simple amphiphiles octadecylamine, which are known to form
micelles in dilute acidic aqueous solution, was also found to form bilayer membranes in
aqueous AgNO3 solution[21]. The aqueous dispersions displayed
regular vesicular morphologies by TEM observation and obvious gel-liquid crystalline phase
transition (Tc = 69 ºC) by DSC measurement. The corresponding cast films showed regular
and sharp XRD patterns[21], from which a slightly tilted tail-to-tail packing
model was inferred. The above features obtained by these conventional characterization
methods of bilayer formation[3] demonstrated that the octadecylamine molecules
formed bilayer membranes through the coordination of their headgroups with Ag+
ions. It was likely that one Ag+ ion coordinated with two octadecylamine
molecules and thus they formed pseudo-double-tailed surfactants with a complexed
headgroup, similar to didodecyldimethylammonium[6]. The metal-coordinated
headgroups induced a parallel arrangement of hydrocarbon chains and reached a balance
between the hydrophilic and hydrophobic interactions as mentioned above.
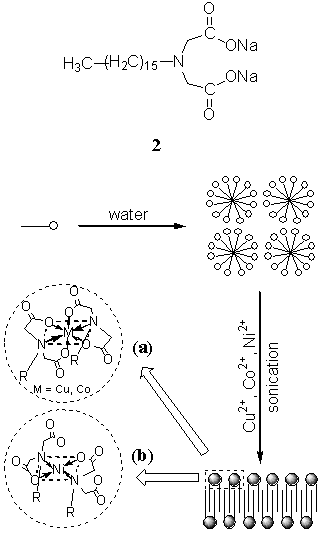
Fig. 3 Schematic representation of micelle formation from 2 in aqueous solution and bilayer formation upon coordination of Cu2+, Co2+, and Ni2+ ions to 2: (a) the proposed coordination model of 2 with Cu2+ and Co2+ ions; (b) the proposed coordination model of 2 with Ni2+ ion.
2.1.2. Organogels in organic solvents
In recent years the studies on the gelation of organic solvents by low molecular weight
organic compounds have been growing into a challenging research field. Some attempts have
been made to correlate the properties of the gels and the structures of the gelators[22,23].
Amphiphiles 3a-c could form stable physical (transparent or translucent) organogels
and gelatinize a number of organic fluids (CCl4, benzene, toluene, o-xylene,
mesitylene, 1,1,2,2-tetrachloroethane, tetrachloroethylene, tetralin, n-octane,
1,1,2-trichlroethane, and cyclohexane) even at a very low concentration[24].
The FTIR spectrum of CCl4 gel of 3b was similar to that of its KBr
pellet, which implied that in the gel the intermolecular hydrogen bonds were formed
between the neighboring molecules 3b close to those in the crystal[24].
The TEM image of the CCl4 gel of 3b showed a number of fibers,
juxtaposed and intertwined by several long slender aggregates with width of ca. 30-100 nm[24].
The corresponding small-angle XRD pattern showed periodical diffraction peaks with the
long spacing of the aggregates (3.43 nm) smaller than twice the evaluated molecular length
of 3b but larger than the fully extended length of one molecule (2.14 nm)[24].
In the combination of FTIR and XRD results, it could be deduced that the gel aggregates
consisted of a repeating bilayer unit, which had a head-to-head packing model with highly
tilted alkyl chains relative to the bilayer normal[24]. Within the bilayer
unit, the amphiphiles were connected by intra- and inter-layer hydrogen bonds to form
hydrogen-bonded networks and developed supramolecular structures[24], which was
schematically illustrated in Fig. 4.
Some structurally related compounds had no gelling ability. The
suitable length of the tail chains was essential for gelation since organogel failed to
form when the tail chain was lengthened to C18 or was substituted by bulky
groups[24]. The racemes of N-dodecanoylalanine did not induce any gelation,
which was similar to that for nonchiral molecule[24]. The finding implied that
the homochiral effect played an important role in gel formation. However, the compounds
containing a bulky group attached to a chiral center did not exhibited gelling ability
probably due to the relatively large steric hindrance that weakened the intermolecular
hydrogen-bonding interaction between the neighboring molecules[24].
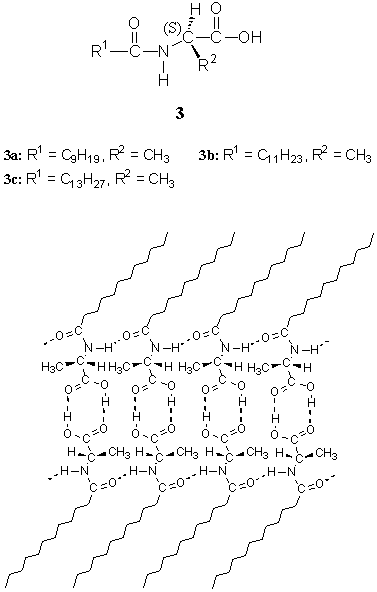
Fig. 4 Schematic representation of the microstructure
of bilayer aggregates of 3b in organogel.
We also found that simple
diamide amphiphiles without chiral center, monoalkyloxamide 4a-c, could form
organogels in a number of organic liquids (CCl4, benzene, toluene, o-xylene,
aceonitrile, aniline, CH2Cl2, 1,2-dichloroethane,
1,1,2-trichlroethane, 1,1,2,2-tetrachloroethane, tetrachloroethylene, and epichlorohydrin)[25].
Similarly, FTIR result of the toluene gel of 4a indicated that in the gel the
primary amide groups of the amphiphiles were connected with each other by intermolecular
hydrogen bonds in the same way as in solid oxamide[25]. The TEM image of
toluene gel of 4a showed a number of fibers, juxtaposed and intertwined by several
long slender aggregates with widths of ca. 30-100 nm[25]. The corresponding
small-angle XRD result indicated that the gel aggregates consisted of a repeating bilayer
unit, which had a head-to-head packing model and highly tilted alkyl chains relative to
the bilayer normal[25]. Within the bilayer unit, the amphiphiles were connected
with intra- and inter-layer hydrogen bonds[25], which was schematically
represented in Fig. 5.
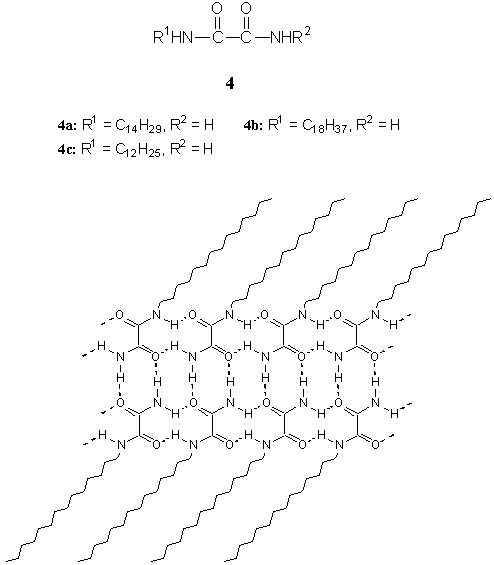
Fig. 5 Schematic representation of the microstructure
of bilayer aggregates of 4a in organogel.
The other structurally
related compounds exhibited no gelling ability. It might be due to the fact that after
substituting one of two hydrogen atoms of the primary amide by an alkyl group, no
interlayer hydrogen bond between the head-to-head arrangements could be formed and the
amphiphiles failed to self-assemble to form ordered bilayer aggregates. Moreover, the
introduction of a long lypophilic chain was essential for gelation since organogel failed
to form when the tail chain was shortened to C4[25].
From the above observations and analyses, it can be concluded that
multiple intermolecular hydrogen bonds and homochiral effects between lypophobic
headgroups and lypophilic chains of suitable length are essential to reach the balance
between the lypophobic interaction and lypophilic interaction for organogel formation. It
is obvious that the novel mechanism of bilayer membranes from simple single-chain
amphiphiles in aqueous solutions is also suitable to that in organic phases to prepare
small-molecule organogels, which will give a guide to studying the structures and
properties of novel elastic materials.
2.2. Monolayer Membranes from Bolaamphiphiles Containing Schiff Base Segments
2.2.1. Monolayer membranes in both aqueous solutions and absolute ethanol
As an extension of single-chain bilayer-forming amphiphiles, the covalent bond connection
of the alkyl chain ends of the bilayer components would produce two-headed amphiphiles,
bolaamphiphiles, which may form monolayer membranes in solvents. Kunitake et al. first
reported the monolayer membranes from such bolaamphiphiles with aromatic rigid segments in
1979[26], but few related studies have been so far reported.
Chart 1
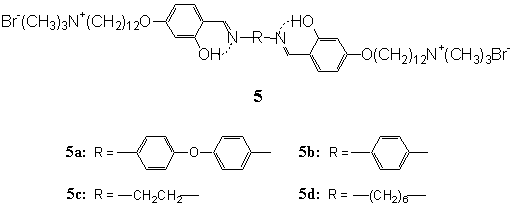
Recently, a series of single-chain bolaamphiphiles containing two salicyidene
groups per molecule connected with varied spacer groups were synthesized (Chart 1).
Bolaamphiphiles 5a- d were readily dissolved in water to give translucent (5a
and 5b) and transparent (5c and 5d) dispersions by sonication[27].
Two types of aggregate morphologies were observed by TEM. Bolaamphiphiles 5a and 5b
with bending and linear rigid segments in the middle of the molecules, respectively,
produced well-developed linear aggregates (tubular and bound fibers), while 5c and 5d
with flexible spacer chains produced regular spherical vesicles[27]. These
observations suggested that the linear aggregate structures could be converted into
vesicle structures by increasing the flexibility of the spacer segments[27].
Temperature-dependent UV-vis spectra showed a transition point for 5a and 5b,
assigned to the conversion of the nonplanar conformations of diphenylazomethine units to
planar ones, and no obvious change for 5c and 5d in the temperature 20-85 ºC[27].
The corresponding DSC measurements displayed single endothermic peak with the enthalpy
change smaller than 10 kJ/mol, considerably lower than those of analogous bilayer
membranes (20-80 kJ/mol), and no detectable transition for 5c and 5d in the
temperature range 15-95 ºC[27]. Combining these results of UV-vis spectra and DSC
measurements, the small enthalpy changes should be attributed to the conversion of
chromophore conformations[27]. NMR spectra of bolaamphiphiles 5a- d in D2O
dispersions at ambient temperature (30 ºC) gave strong signal of N-methyl groups, relatively narrow signals
of methylene groups, and no discernible signal of Schiff base units[27]. These
signals became narrower and stronger at elevated temperatures, and the narrower and
stronger signals of Schiff base units were observed at 80 ºC[27]. These
indicated that the headgroups in water had high mobility, and the alkyl chains were less
restricted and packed loosely to each other in the liquid-crystalline state[27].
The bis-Schiff base segments stacked densely through p-p interaction between them was the driving force for monolayer
formation. FTIR spectroscopy provided further evidence for the formation of the
liquid-crystalline monolayer membranes, and no marked gel-liquid crystalline phase
transition was observed in the temperature range 0-100 ºC[27].
In absolute ethanol, 5b-d gave translucent dispersions upon
sonication at room temperature[28]. 5b formed irregular vesicles, and 5c
and 5d mainly displayed ellipsoidal vesicles. The UV-vis spectrum of 5b in
ethanol (366 nm) was close to that in water (369 nm) in contrast to the molecularly
dispersed species in DMSO (382 nm)[28]. These indicated that bolaamphiphiles 5b-d
formed monolayer membranes in absolute ethanol and the spacer groups between the Schiff
base segments had an influence on aggregate morphologies[28]. The strong p-p interaction between the bis-Schiff
base segments played an important role in molecular assemblies.
2.2.2. Monolayer membranes in organic solvents
When the two hydrophilic headgroups of bolaamphiphiles 5c were substituted with two
hydrophobic methyl groups, bolaamphiphile 5c became long-chain bis-Schiff base
derivative 6. The 6 and its metal complexes 6(GaMe2)2
and 6(InMe2)2 were readily dispersed in organic solvents of
low polarity, such as, chloroform, benzene, toluene, and so on, to give transparent and
translucent solutions[29]. The dispersion of 6 in chloroform displayed
typical vesicular morphologies, the dispersion 6(GaMe2)2
exhibited linearly connected vesicles, and the dispersion 6(InMe2)2
showed hexagonally perforated membrane structure[29]. Their cast films
displayed periodic XRD patterns. These demonstrated that 6 and its metal complexes
formed stable monolayer membranes in the organic solvents. 1H NMR spectra of 6
and its metal complexes in CDCl3 showed line broadening to some degree[29].
The signals of metal-coordinated Schiff base segments were weak and broad, while the peaks
of the corresponding alkyl chains were strong and sharp. The similar conclusions could be
drawn: they formed liquid-crystalline monolayer membranes in the organic solvents of low
polarity (see Fig. 6).
The zinc complex (1:1) of 6 also gave translucent emulsion in
these organic solvents by shaking, however, TEM observation showed uniform lamellar
morphologies in chloroform, which was different from the vesicular morphologies of 6
and its two metal complexes described above[30]. The layer spacing of the zinc
complex cast film (2.78 nm) was close to that of the cast film of 6 (2.64 nm) and
smaller than the evaluated molecular length for 6[30]. These indicated
that the organized monolayer membranes were formed by zinc complex in organic solvents.
Taking into account the lamellar aggregate morphologies and the melting temperature 320 ºC, the zinc
complex was deduced to be a coordination polymer[30], which depressed the bend
of methylene spacers and displayed linear aggregates. The complex structure was further
supported by the fact that zinc complex had stronger fluorescence intensity than 5
since the rigidity of the coordination polymer reduced the loss of energy via vibrational
motions[30]. Furthermore, FTIR, NMR, and DSC measurements demonstrated that the
monolayer membranes of zinc complex in organic solvents were in the liquid-crystalline
state, which was schematically illustrated in Fig. 7. The bis-Schiff base and coordinated
bis-Schiff base segments constituted the ordered stacking backbones of the monolayer
membranes as solvophobic parts, and the alkyl chains acted as solvophilic parts in the
organic media[30].
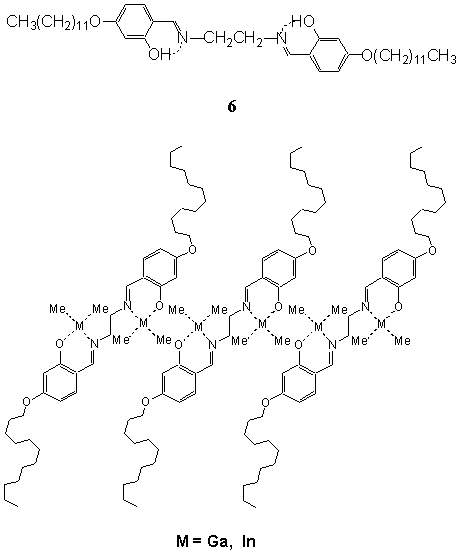
Fig. 6 Schematic illustration of the monolayers of
organometallic complexes of 6 in organic solvents.
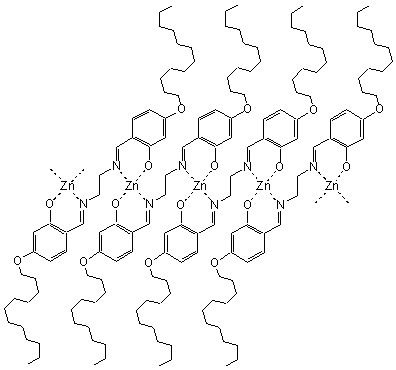
Fig. 7 Schematic illustration of the monolayer of zinc coordination polymer of 6 in organic solvents.
It is obvious that the
organized molecular assemblies of different functions may be prepared from well-designed
amphiphiles in their broad sense in various kinds of media with different driving forces:
(i) from conventional amphiphiles to those in their broad sense; (ii) from molecular
assemblies in aqueous solutions to those in organic solvents; (iii) from basic research to
applied science. These indicate that the broad filed of molecular assemblies is to be
further studied.
2.3. LB Films of Long-chain Schiff Base Derivatives
Schiff base compounds and their metal complexes exhibit very interesting properties and
have been well characterized since the late 1920s, however, their applications are
restricted due to their poor stability. It has been shown that their stability and
properties are greatly increased after the introduction of the chromophores into ordered
Langmuir monolayers and LB films[31]. The novel amphiphile
2,4-dihydroxy-N-octadecylbenzylideneamine 7 could form stable monolayers at the
air-water interface, which underwent a partial conversion from intermolecular to
intramolecular hydrogen bonds as well as the change in the Schiff base headgroup
orientation from the "flat-on" to "end-on" structures around the surface pressure 30-40 mN/m upon compression[32,33].
Both intermolecular and intramolecular hydrogen bonds existed in the LB films depending on
deposition pressure[34]. The transferred LB films were Z type below 30 mN/m
with a preference of intermolecular hydrogen bond and Y type above 40 mN/m with a
preference of intramolecular hydrogen bond[34]. At the surface pressures 5-30
mN/m, the Tc of the LB films was at approximately 60 ºC, when the surface
pressure was increased to 45 mN/m, the Tc was decreased to about 45 ºC[34].
That demonstrated the occurrence of the partial conversion from intermolecular to
intramolecular hydrogen bonds around 30-40 mN/m, due to the tautomeric structures of the
hydrophilic headgroups (Chart 2).
Photoinduced proton transfer from one moiety to the other in one
molecule is of great interest. It is regarded as one of the simplest chemical reactions
and is related to some biological processes. The Schiff base moieties in the LB film of 7
deposited at 40 mN/m, with the zwitterions configuration as a result of intramolecular
hydrogen bonds, underwent a photoisomerization process to a keto configuration after 1-h
ultraviolet (UV) irradiation[35]. It would be reasonable to expect a proton
transfer followed a rotation of a phenylene ring in the process of photoisomerization that
would induce a gradual expansion of the LB film and perhaps a partial rearrangement of the
Schiff base[35]. We found that equimolar mixture of 7 and (Et4N)2[Zn(dmit)2]
(H2dmit = 4,5-dimercapto-1,3-dithiole-2-thione) could form a typical condensed
monolayer at the air- water interface[36]. 7 was a weak electron donor,
and (Et4N)2[Zn(dmit)2] with a transition metal and
sulfur-containing ligands was a typical electron acceptor. Their equimolar mixture in
chloroform solution indicated little charge transfer between them, but the magnitude of
charge transfer was dramatically increased after iodine doping[36]. The similar
phenomenon of charge transfer was observed for the LB film of equimolar mixture after
iodine doping[36]. This will provide a new and simple way to obtain organic
materials with high electrical conductivity.
Chart 2
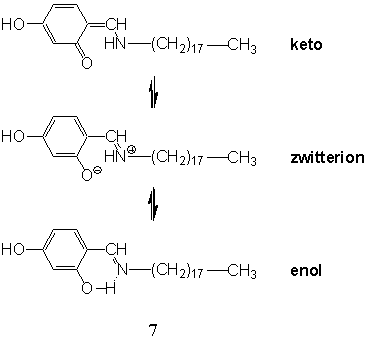
The LB films of copper complexes of 7
could be prepared through three different ways. The monolayers and LB films of
presynthesized copper complex were referred to as surface films, and the monolayers and LB
films from the spreading of 7 on aqueous Cu(CH3COO)2
solutions were described as subphase films. There was a little difference in p-A isotherm between the surface film
and the subphase film, but their LB films showed very similar FTIR spectra[32].
The alkyl chains in the two LB films took a biaxial orientation at the tilt angle 22-24°
from the film normal, in contrast to a uniaxial orientation in the LB film of 7 at
the tilt angle 30° [51]. That implied that the subphase film possessed a
similar molecular structure to the surface film. Another type of copper complexes was
described as exchange film, which was formed after Cu2+ ions penetrated into
the LB film of 7[37]. The exchange film showed different FTIR spectral
profile from the surface film and subphase film, suggesting that the exchange film might
have a different molecular structure. The alkyl chains in the exchange film were
uniaxially oriented at the angle 35° from the film normal[37]. The biaxial
orientation of the alkyl chains in the surface film and subphase film were considered to
be due to a close-packed superlattice network of the complexed headgroups that were
subjected to the shear forces induced during film transfer[37], while no
transfer-induced orientation effect occurred for the exchange film, the formation of which
was controlled by the molecular structure of the LB film of 7, such as, headgroup
orientation and chain packing. This might be the reason for the difference in molecular
structure between the exchange film and other complex films.
Accordingly, the three types of complex films showed different phase
behaviors. The surface film and subphase film exhibited two phase processes at
approximately 50 and 110 ºC, respectively[32], however, small difference in phase
behavior was also observed for the two films, which was consistent with the difference in
monolayer behavior[32]. The exchange film displayed one phase process around
the transition temperature 70 ºC[37], similar to the phase behavior of LB film of 7.
It is clearly seen that the phase behaviors of the complex films are closely related to
their molecular structures.
Calixarenes are versatile macrocyclic molecules with a number of
applications[38] and can form a perfect monolayer at the air-water interface.
However, the monolayers of calixarenes could not be transferred onto solid substrates
owing to their strong rigid properties. The introduction of the copper complex of 7
into p-tert-butylcalix[4]arene resulted in a successful transfer of multilayer LB films
from the mixed monolayers at the air-water interface[39]. The interfacial
behavior and thermodynamic estimation of the mixed monolayers indicated that a strong
intermolecular interaction existed between the two components, and the further studies of
FTIR and UV-vis spectra demonstrated that the intermolecular interaction resulted from the
interaction between the headgroups of the two components[39].
2.4. LB Films of Conjugated Polymer Derivatives
Conjugated polymers have been subject of great interest from both fundamental point of
view and applied science perspective. Poly(p-phenylenevinylene) (PPV) derivatives stand
out for the ability to combine a reasonably good environmental stability and high
optoelectronic properties[40], and are viewed as active components for organic
electroluminescent devices.
LB deposition technique in molecular processing has represented a
powerful method for organizing rod-like polymers. PPV derivative (MH-PPV) 8 (Chart
3) could form a stable monolayer at the air- water interface, and the monolayer could be
transferred to fabricate Y-type LB films by the vertical method[41]. The UV-vis
absorption of the conjugated polymers from the p-p*
transition is very sensitive to the direction of polarization of an incident light. It was
shown that the backbones of 8 were preferentially aligned along the dipping
direction in the LB film[41]. Optical band gap is defined as the lowest energy
band edge of the solid-state electronic spectra of conjugated polymers. An optical band
gap of 2.11 eV was obtained for the LB film of 8[41], which was slightly
smaller than that of PPV (~ 2.5 eV). The results indicated that the ordered arrangement of
8 in the LB film could reduce its optical band gap. In the LB film, the conjugated
backbones were aligned in a side-by-side parallel fashion and packed with the plane of its
p system approximately perpendicular to the substrate surface (H aggregate), and the long
chains presented all-trans conformation at a tilt angle of 25° from the film normal
obtained from small-angle XRD pattern and polarized FTIR spectra[41]. The
atomic force microscope (AFM) study of the LB monolayer further indicated that the
conjugated backbones of 8 were arranged in 2D ordered array with the elongated
domain parallel to the dipping direction in the monolayer[42].
Chart 3
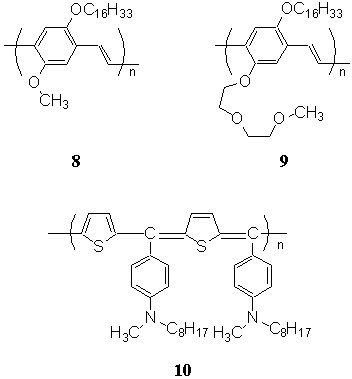
When the hydrophilic headgroup of 8
was substituted by di(ethylene oxide) ethyl ether, 8 was changed to 9
(MMDU-PPV). The monolayer of 9 at the air-water interface showed three kinks of
aggregation states in the process of compression and could be transferred to fabricate
Z-type LB films[43]. The polarized UV-vis spectrum of the LB film deposited at
the surface pressure 14 mN/m indicated that the conjugated backbones were preferentially
aligned parallel to the dipping direction, while the polarized spectrum of the LB film
deposited at 30 mN/m indicated that the conjugated backbones were isotropic, which was due
to the collapse of the monolayer in this case[43]. Polarized FTIR result
indicated that the alkyl chains of 9 in the LB film were further tilted in
comparison with those of 8[43]. The optical band gap of 9 in the
LB film was 1.8 eV[43], smaller than that of parent PPV (~ 2.5 eV).
Poly(heteroarylene methines) is known as the small-bandgap conducting
polymer in all kinds of conjugated polymers[44]. Its derivative 10
(PTABQ) could be successfully spread onto water surface[45]. The behavior of
the monolayer of 10 at the air- water interface was sensitive to its spreading
solvent. Chloroform solvent gave an inhomogeneous film (type A), and less evasive solvent
o-xylene yielded a homogeneous film (type B)[45]. A water-soluble amphiphile as
an extractable spread-aiding component was introduced to the chloroform solution of 10,
a flexible and homogeneous film (type C) was produced after dedocanol gradually detached
from the film into subphase upon compression[45]. The AFM images of the type C
film was the smoothest among three ones, and FTIR studies showed that the alkyl chains in
the type C LB film were more ordered than the others[45]. The optical band gap
of 10 in the LB film was 1.38 eV[45], which was much smaller than that
of related poly(heteroarylene methine) derivatives (1.79- 1.94 eV)[46]. It was
considered that the ordered arrangement of 10 in the LB films might improve the
conjugation of the backbones and the p - p interaction between them reduced the optical
band gap of 10[45]. The type C LB film had an electrical conductivity of
8.1×10-8 S/cm, and the electrical conductivity was increased to 5.7×10-7
S/cm when the LB film was doped with iodine vapor[45], however, the electrical
conductivity was still low. It was attributed to two parts: (i) the low molecular weight
and bulky side chains of 10, which made p electrons difficult to delocalize in the
short conjugated backbones; (ii) the low level of doping in the LB film[45].
3. BIOLOGICAL MOLECULAR ASSEMBLIES
3.1. LB Films of N-Acyl Amino Acid
Over the last decade, considerable attention has been drawn toward the studies of
N-acyl amino acid since amino acids are known to play an important role in membrane
sections[47-51]. The N-acyl amino acid monolayers at the air- water interface
and LB films have been studied through fluorescence microscopy[47,48] and FTIR
spectroscopy[49-57]. The FTIR studies of the LB films of
N-octadecanoyl-L-alanine 11 indicated that the enantiomeric molecules assembled regularly
through an extended intermolecular hydrogen-bonded network and twisted from neighbor to
neighbor to develop an anisotropic aggregate in the 2D crystalline array[52-54].
The intermolecular hydrogen-bonding interactions and homochiral effects between the
hydrophilic headgroups strengthened the interactions between the hydrophobic chains, so
that the Tc of the LB films of 11 (122 ºC)[52,53] was
almost twice as high as that of LB films of same-chain-length stearic acid (65 ºC)[58].
It was shown that the presence of metal ions in the aqueous subphases might give rise to
considerable compression or expansion of the film-forming molecules as well as to an
increase or decrease in chiral discrimination in the respective monolayers depending on
kind of metal ion[50,51,55]. The condensing effects of Ag+, Zn2+,
and Pb2+ ions led to an increase in the intermolecular hydrogen-bonding
interaction between neighboring molecules 11[55,57]. A preference of chelating
bidentates was formed between metal ions and carboxylate groups in the cases of Ag+
and Zn2+ ions, and the alkyl chains in silver and zinc complex LB films took a
biaxial orientation[55]. FTIR results indicated that two types of metal
complexes were formed with different headgroup complexes and chain orientation in the case
of Pb2+ ions: (i) the chelating bidentates were formed between Pb2+
ions and carboxylate groups, and the C-C-C planes of the corresponding chains were
favorably oriented parallel to the substrate surface; (ii) the bridging bidentates were
formed, and the C-C-C planes were preferentially oriented perpendicular to the substrate
surface[57] (see Fig. 8). Whereas, the expanding effects of Ca2+, Cd2+,
Ni2+, and La3+ ions resulted in the formation of intramolecular
hydrogen bonds via a five-membered ring structure between the NH bond of amide group and
the carbonyl group of carboxylate[55]. A preference of hydrogen-bonded
monodentated or unsymmetric ionic complexes was formed between these metal ions and
carboxylate groups, and the alkyl chains in these LB films were uniaxially oriented at the
angle 40-50° with regard to the film normal[55] (Fig. 8) .
The differences in the film structure would give rise to a change in
the basic physicochemical properties of these LB films. It was found that metal complex
and hydrogen bond played important roles not only in film structure but also in film phase
behavior. The LB films of silver and zinc complexes showed the phase behavior of glass
transition, especially for the zinc complex LB film[55]. This was owing to a
gradual dissociation of the enhanced intermolecular hydrogen-bonding interaction over a
wide temperature range[55], in contrast to a complete dissociation of the
relatively weak intermolecular hydrogen-bonding interaction in the LB films of 11
around the Tc (122 ºC). The interchain interaction, induced by the increased
intermolecular hydrogen-bonding interaction, played a determinative role in phase process
of the LB films. Calcium, cadmium, and nickel complex LB films exhibited the phase
behavior of thermotropic liquid crystal more than one transition process[55].
The five-membered ring structures of intramolecular hydrogen bonds containing chiral
carbons might act as rigid or semirigid groups and had the similar effects to the
mesogenic units in usual liquid crystals[55]. The phase behaviors of these
metal complex LB films showed a clear dependence of molecular structure of hydrogen bond
and complex formation.
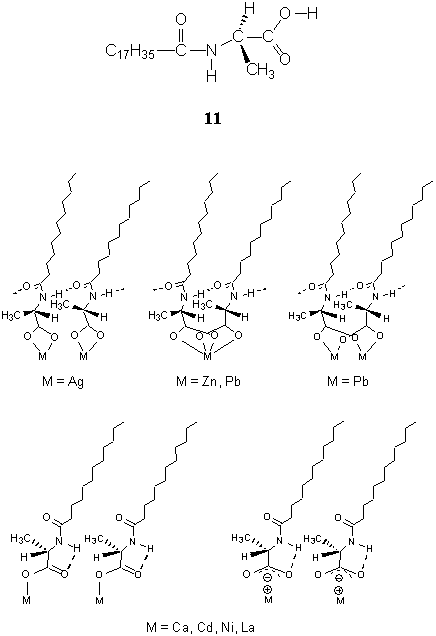
Fig. 8 Probable structures of the monolayers of metal
N-octadecanoyl-L-alaninate.
LB films usually possess a
highly crystalline lamellar structure and consist of a plurality of domains with diameters
between 0.1 and 10 m m[59], and the distributed defects among them may provide
transport channels for access of water-soluble materials, that is, metal ions, to LB film
matrixes[60,61]. Ag+, Zn2+, Pb2+, Ca2+,
Cd2+, and Ni2+ ions penetrating into the LB films of 11 were
selectively exchanged with the carboxylic acid groups at their self-buffered pH values[56,57].
The behaviors were different from the interactions between the corresponding monolayers at
the air- water interface and the metal ions in the aqueous subphases. The ion exchange in
the LB films was controlled by the film structure of the intermolecular hydrogen-bonded
network[56]. Ion exchange was favorable to Ag+, Zn2+, Pb2+
ions, as these ions promoted an increase in intermolecular hydrogen-bonding interaction
and a decrease in intermolecular distance[56,57]. Ag+ ions gave rise
to a partial ion exchange because the intermolecular hydrogen-bonding interaction was only
slightly increased and the conformations and packing of the alkyl chains remained
unchanged[56]. Zn2+ and Pb2+ ions could lead to a
complete ion exchange after 5 and 29 h, respectively[56,57]. Although the
intermolecular hydrogen-bonding interaction was significantly enhanced in the case of Pb2+
ions, the almost orthogonal orientation of the C-C-C planes of the two types of metal
complexes might be the reason for the slow ion exchange within the LB film[57].
Ion exchange was unfavorable to Ca2+, Cd2+, and Ni2+
ions, as it was suppressed by the occurrence of intramolecular hydrogen bonds and the
increase of intermolecular distance[56]. However, Ca2+, Cd2+,
and Ba2+ ions at pH 7.0 could penetrate into stearic acid LB films through
structural defects and reacted with carboxylic acid groups in the films[62].
The ion-exchange reactions were complete for Ca2+ and Cd2+ ions
within a few minutes[62]. The reverse exchange processes in the LB films of
metal complexes by protons were consistent with the above results. Ion exchange was
unfavorable to zinc and silver complexes as it was suppressed by the weakening of
intermolecular hydrogen-bonding interactions and the increase of intermolecular distance,
and it was extremely favorable to calcium, cadmium, and nickel complexes in converting
from intramolecular to intermolecular hydrogen bonds within a couple seconds [38]. The
molecular assemblies in the LB film exhibited a different performance from single
film-forming molecules.
3.2 LB Films of Nucleolipid Amphiphiles
Many biological molecules are found to have specific molecular recognition properties to
present various biological functions. The most notable example of biological recognition
is complementary base pairing in nucleic acids. The mutual recognition of Watson-Crick
complementary bases in nucleic acids by means of multiple hydrogen bonds is the most
efficient mechanism of accumulating, storing, reproducing, and evolving genetic
information. Base-base interactions might be driven by p-p stacking and/or intermolecular
hydrogen-bonding interactions, both necessary for nucleic acid stabilization, but it is
worthwhile to recall that the hydrogen-bonding interaction does not provide significant
driving force for molecular recognition in bulk water due to the strong competitive
binding of water molecules. The Langmuir monolayers at the air-water interface provide unique environments for molecular
interactions and consequently for molecular recognition[2,63-69]. Bases in DNA
interact mainly via hydrogen bonds with their Watson-Crick partners on the complementary strand, and each nucleic acid
strand is greatly stabilized by stacking interactions between neighboring bases. The
organized nucleolipid monolayers at the air-water
interface might act as single nucleic acid strands, due to the hydrophobic interaction
between the corresponding alkyl chains, to recognize complementary bases in the subphase,
which not only provide more information on molecular recognition of nucleic acid bases in
nature but also is suitable to being studied with molecular spectroscopy using LB
technique.
Chart 4
3.2.1 Molecular recognition between adenine
and thymine/uracil moieties
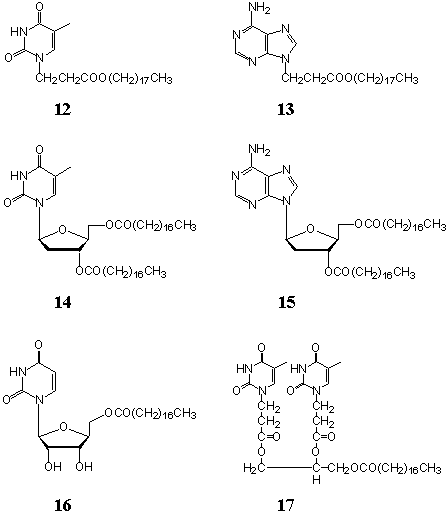
A series of nucleolipid amphiphiles with base moieties as headgroups were synthesized[67]
(Chart 4). All of them could form stable monolayers at the air-water interface on pure water and complementary base-containing
subphases. It is known that thymidine bases in aqueous solutions at room temperature could
not dimerize upon UV irradiation, however, the photodimerization of thymine moieties were
reported to generate upon UV irradiation after its solution was frozen[68]
because under this condition the molecular arrangement became more regular compared with
that in the room-temperature solution. The LB film of nucleolipid 12 transferred
from pure water gave an absorption maximum of 270 nm. The intensity decreases gradually
upon UV irradiation and disappeared after 5 h, which indicated that the photodimerization
of the thymine moieties in the LB film took place at room temperature[69]. The
photoproduct of thymine bases in DNA upon UV irradiation had the cyclobutane-type
structure, whose formation could be followed by the variation of 270-nm band in the UV
absorption spectra. In the LB film matrix, the constituent molecules were arranged
regularly and close to each other, which facilitated the photodimerization of the thymine
moieties in the headgroups. The photoproduct cyclobutane was formed only when the thymine
moieties must be able to come into close contact. From crystallographic data, the maximum
distance between the double bonds of the thymine rings for dimerization should be at most
0.42nm[70]. When the LB films of 12 were transferred from aqueous
adenosine solutions (1 and 5 mM), the maximum absorption intensity increased with
adenosine concentration[71], which suggested the occurrence of molecular
recognition between thymine and adenosine moieties through intermolecular hydrogen-bonding
interaction (see Fig. 9). Similarly, the two LB films also underwent photodimerization
upon UV irradiation[71]. On the other hand, this indicated that the regular
arrangement of the monolayers with thymine moieties could recognize their complementary
bases in the subphases. FTIR spectroscopic studies indicated that prior to molecular
recognition the alkyl chains in the LB film were biaxially oriented and both the chains
and thymine planes were considerably tilted from the film normal[72]. The tilt
angles were further increased after molecular recognition. The Tc of the LB
film after molecular recognition was reduced in comparison with that prior to molecular
recognition[72].
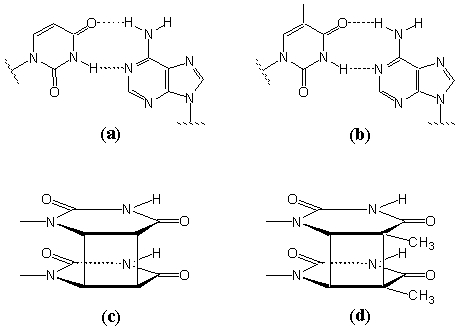
Fig. 9 Schematic illustration of complementary multiple hydrogen bonds between adenine and uracil groups (a) and between adenine and thymine groups (b), and of photodimerization between uracil groups (c) and between thymine groups (d).
For nucleolipid 13 with
adenine moiety, the corresponding LB films transferred from aqueous thymidine and uridine
solutions, respectively, could undergo similar photodimerization upon UV irradiation[73].
As mentioned above, no photodimerization took place in aqueous thymidine and uridine
solutions at room temperature due to the random distribution of the molecules in the
solutions. The occurrence of photodimerization in the LB films were attributed to the
regular and close arrangement of the complementary bases thymidine and uridine induced by
the molecular recognition of adenine moieties in the monolayers at the air-water interface
through intermolecular hydrogen-bonding interaction. This was a powerful UV spectroscopic
evidence for molecular recognition between complementary base pairs adenine and
thymidine/uridine. However, the time of photodimerization reaction was different, 6 h for
the LB film containing thymidine bases and 4 h for the LB film containing uridine bases[73].
The difference was considered to result from the steric hindrance caused by the methyl
groups in the thymine moieties. The uracil moieties were likely to be more regular and
closer to each other in the LB film than the thymine moieties[73]. The alkyl
chains in the LB film containing thymidine bases were biaxially oriented, while the chains
in the LB film containing uridine bases were uniaxially oriented, similar to the chains in
the LB film transferred from pure water[74]. The discrepancy in chain
orientation was likely to result from the steric effect caused by the methyl groups of the
thymine moieties. However, the order-disorder phase transition of these LB films
transferred from various subphases showed no significant difference[74].
For nucleolipids 14 (single headgroup (T)-double chains), 16
(single headgroup (U)-single chain), and 17 (double headgroups (T,T)-single chain),
the collapse surface pressure of the monolayers at the air- water interface on pure water[75,76]
was in the order of 14 > 16 > 17. Considering the sizes and
hydrophilicity of the base moieties and the packing density of the alkyl chains in these
nucleolipid molecules, the interaction balance between hydrophilic headgroups and
hydrophobic chains is in the order of 14 > 16 > 17, which is
consistent with the order of the collapse pressure of the corresponding monolayers. All of
the LB films transferred from pure water underwent photodimerization upon UV irradiation[75,76],
but their photodimerization rate is basically in the order of 14 < 16
< 17. The difference could result from the molecular structure of three
nucleolipids, i.e., the numbers of hydrophilic headgroups and hydrophobic alkyl chains.
The photodimerization reaction is known to depend on the double-bond distance of adjacent
base moieties. It is obvious that the distance of the thymidine moieties in the monolayers
of 14 (double chains) is larger than that of the uridine moieties in the monolayers
of 16 (single chain) besides the factor of the steric hindrance from the thymine
moieties. For nucleolipid 17 (double headgroups), the molecular packing density in
the corresponding monolayers might be lowest, however, two thymine moieties in one
molecule were close-packed to each other. The distance between them was small enough for
the occurrence of photodimerization although there might be steric hindrance from the
methyl groups in the thymine moieties. Some conclusions can be drawn from the comparison
of the photodimerizaton reactions in the LB films prior to and after molecular
recognition: (i) the photodimerization rate is reduced after molecular recognition; (ii)
the magnitude of rate reduction after molecular recognition is relevant to recognition
capacity of complementary base pairs, i.e., the stronger the recognition capacity (the
more the adsorbed complementary bases), the more the photodimerization rate is reduced.
The occurrence of molecular recognition can give rise to a decrease in packing density of
thymine/uracil moieties in the monolayers as well as a change of base ring orientation.
FTIR spectroscopic studies also showed that the molecular arrangement of the alkyl chains
in the LB films after molecular recognition was less dense that that prior to molecular
recognition[76].
Surface-enhanced Raman scattering (SERS) technique was used to study
the above molecular recognition systems for the first time[77]. High-quality
SERS spectra were obtained to give much rich information on molecular recognition of
complementary base pairs. The SERS spectra of the molecular recognition systems could be
classified into two groups: one contained nucleolipids 12, 14, 16,
and 17 with thymine/uracil moieties; the other contained nucleolipids 13 and
15 with adenine moieties[77]. The SERS spectra of the LB monolayers
transferred from pure water were greatly different from the normal Raman spectra of the
corresponding power samples. The vibrational bands relevant to long-chain hydrocarbon
could not be observed in the SERS spectra[77]. Some of vibrational bands
relevant to the base moieties were observed in the SERS spectra, however the corresponding
ring-breathing vibrational bands were weak or disappeared because the strong interactions
between the base rings and silver surfaces restricted the motion of the rings, which were
almost lying flat on the silver surface[71]. For the LB monolayers transferred
from complementary base-containing subphases, all of the above bands were decreased in
intensity or disappeared, and new enhanced bands were observed due to the ring skeletal
vibrations and ring breathing vibration of the complementary bases[77]. These
changes indicated that the nucleolipid monolayers at the air-water interface were capable
of interacting with dissolved complementary bases in the subphases, and the complementary
bases were also transferred onto solid substrates along with the monolayers. The
complementary base rings probably took an end-on structure on the silver surface[71].
For second group, the SERS spectra of the LB films transferred from aqueous thymidine
solutions were similar to those transferred from aqueous uridine solutions, but the
enhanced effect of the thymine moieties was much weaker than that of the uracil moieties
probably due to the steric hindrance caused by the methyl groups in the thymine moieties[77].
SERS spectroscopy is demonstrated to be very sensitive and supplies more information about
the molecular recognition of nucleic acid base pairs.
3.2.2 Molecular recognition between guanine and cytosine moieties
The nucleolipids 18 and 19 containing cytosine and guanine moieties
respectively (Chart 5) could form stable monolayers on pure water and complementary
base-containing subphases. The corresponding monolayers were transferred onto solid
substrates to be Z-type LB films[78,79]. FTIR spectroscopic evidence for
molecular recognition of 18 to its complementary base guanosine was obviously
observed from the frequency downshift and width increase of the NH2 stretching
vibrational band[78]. The triple hydrogen-bonding pattern of the molecular
recognition is shown in the inset of Fig. 10. UV-vis, FTIR, and SERS spectral studies
indicated that the cytosine rings in the monolayers of 18 underwent a change in
orientation and aromatic stacking from the flat-on geometry without stacking interaction
before molecular recognition to an end-on one in the J-aggregate fashion after molecular
recognition[78], which is schematically shown in Fig. 10. The LB film of 18
transferred from pure water showed a crystal transition at the temperature 145 ºC[78].
The high Tc of the LB film resulted from the intermolecular hydrogen-bonding
interaction between the adjacent cytosine headgroups (Fig. 10). which was the driving
force for the cytosine rings to orient favorably parallel to the film surface without
aromatic stacking at room temperature. At elevated temperatures, the intermolecular
hydrogen-bonding interaction was progressively weakened with temperature, and the planes
of the cytosine rings gradually lay flat on the surface (Fig. 10)[78]. The LB
film of 18 transferred from guanosine-containing subphase (after molecular
recognition) displayed a surprising phase transition behavior. It was shown that the
intermolecular hydrogen-bonding interaction between the cytosine and guanosine base pairs
was stable below 60 ºC[78]. Although the intermolecular hydrogen-bonding
interaction between the cytosine and guanosine base pairs was stronger than that between
the cytosine moieties in the monolayers, the hydrogen-bonding interaction between the base
pairs after molecular recognition could not strengthen the interaction between the
hydrophilic headgroups or the interaction between the corresponding hydrophobic chains.
When the temperature was elevated above 60 ºC, the gauche conformers in the alkyl chains was increased, and the
triple hydrogen-bonding interaction started weakening, at the same time, the
hydrogen-bonding interaction between the adjacent cytosine moieties in the monolayers was
formed gradually[78]. After the temperature was increased to 120 ºC, the
hydrogen-bonding interaction between the cytosine and guanosine base pairs was completely
dissociated, and the hydrogen-bonded network between the adjacent cytosine moieties in the
monolayers was formed and the almost all-trans conformation in the alkyl chains was
recovered, accompanied with the change of the cytosine ring orientation from the end-on
fashion to a flat-on one[78]. The above phase transition behavior not only
provided powerful evidence for molecular recognition of the cytosine and guanosine base
pairs at the air-water interface through strong
hydrogen-bonding interaction but also revealed the process of the molecular recognition in
all directions (Fig. 10).
Chart 5
It is known that guanines possess a unique ability to undergo
spontaneous formation of a regular, ordered quadruplex structure in aqueous solution[80,81].
When a hydrophobic long chain was attached to the nitrogen at N (7) in the guanine ring
like 19, the formation of quadruplex structure between guanine moieties is
depressed. The FTIR spectrum of the LB film of 19 transferred from
cytosine-containing subphase showed a downshift of the C=O stretching band relevant to
guanine moieties and the characteristic bands of cytosine moieties[79]. These
spectral changes indicated the occurrence of molecular recognition of 19 to its
complementary base. The polarized FTIR spectral studies indicated that the alkyl chains in
the LB film after molecular recognition were further tilted away from the film normal in
comparison with those before molecular recognition[79]. The alkyl chains in the
LB film prior to molecular recognition were melted in a wide temperature range from 70 to
150 ºC[82], which might suggest that the guanine headgroups
were involved in an intermolecular hydrogen-bonded pattern, while the chains after
molecular recognition were melted in the temperature range from 50 to 110 ºC[82].
The reduced thermal stability reflects the occurrence of molecular recognition between the
complementary base pairs. The phase transition behaviors of LB films of 18 before
and after molecular recognition are different from those of 19 indicate the
difference in hydrogen-bonding interaction between guanine-guanine moieties vs.
cytosine-cytosine moieties in the monolayers.
FTIR, UV-vis, and SERS spectroscopic techniques provide rich
information on molecular recognition between the complementary base pairs in the LB films.
To obtain direct information about molecular recognition under biological environments,
in-situ spectroscopic studies of the monolayers at the air-water interface are necessary.
The molecular recognition of the monolayers of 18 at the air- water interface on
the guanosine-containing subphases (H2O/D2O) was studied using
infrared reflection absorption (IRRAS) technique[83]. The alternate spectral
collection of sample and reference troughs and the use of D2O subphase can get
rid of interference from water vapor and shift the absorption bands of water away from
interested frequency regions. In the case of guanosine/D2O subphase, the
spectral changes of three absorption bands (NH2 bending, C(2)=O stretching,
C(4)=N(3) stretching) corresponded to the formation of triple intermolecular hydrogen
bonds was clearly observed using IRRAS technique[83]. The spectral results
indicated that the cytosine rings took a change in orientation from the flat-on fashion
before molecular recognition to an end-on one after molecular recognition[83].
The theoretical simulation based on the spectral results showed that the alkyl chains in
the monolayers at the air-water interface underwent a orientation change from 20° with
respect to the film normal before molecular recognition to 30° after molecular
recognition[83]. The change reflects that the interaction between hydrophilic
headgroups can influence the interaction between hydrophobic alkyl chains.
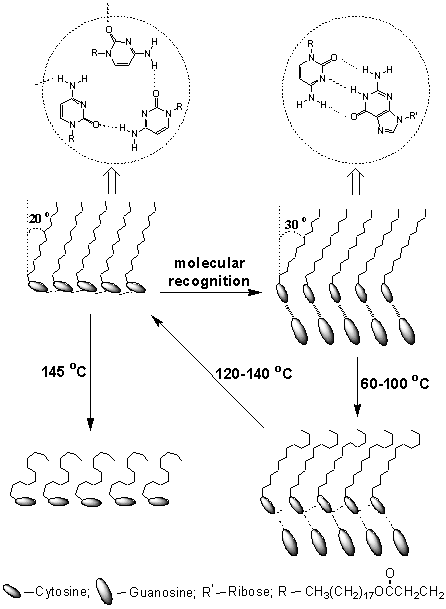
Fig. 10 Schematic illustration of the hydrogen-bonding
patterns, ring and chain orientation, and the corresponding phase transition of 18 in
the monolayers and LB films prior to after molecular recognition.
4. CONCLUDING REMARKS AND PROSPECTS
The studies of organized molecular assemblies based on the self-organization and molecular
recognition of cell membranes are helpful for understanding structures and functions of
biomembranes and for preparing functional molecular assemblies. Building up the
relationship between the structures and performances of molecular assemblies is based on
the understanding of the structure of single molecules and molecular assemblies.
Well-designed amphiphiles are crucial to achieving molecular assemblies of various
functions. New and sensitive spectroscopic techniques are important to giving a deep
insight into the relationship between the structures and performances of molecular
assemblies. Molecular Dynamics has been recently applied in simulation of molecular
aggregates. Spectroscopic characterization combined with theoretical calculation will be
very helpful in the elucidation of the microstructures and properties of molecular
aggregates.
REFERENCES
[1] Ringsdorf H. Supramol. Sci., 1994, 1: 5.
[2] Ringsdorf H, Schlarb B, Venzmer J. Angew. Chem. Int. Ed. Engl., 1988, 27:113.
[3] Kunitake T. Angew. Chem. Int. Ed. Engl., 1992, 31: 709.
[4] Fuhrhop J H, Helfrich W. Chem. Rev., 1993, 93: 1565.
[5] Liang Y, Zhang Z. J. Dispersion Sci. Technol., 1995, 16: 339.
[6] Kunitake T, Okahata Y. J. Am. Chem. Soc., 1977, 99: 3860.
[7] Li G, Tian Y, Liang Y. J. Chem. Soc., Chem. Commun., 1990, 889.
[8] Li G, Liang Y. J. Chem. Soc., Chem. Commun., 1991, 1773.
[9] Li G, Tian Y, Liang Y. J. Chem. Soc., Faraday Trans., 1993, 89: 1365.
[10] Liang Y, Wu L, Tian Y et al. J. Colloid Interface Sci., 1996, 178: 703.
[11] Liang Y, Zhang Z, Wu L et al. J. Colloid Interface Sci., 1996, 178: 714.
[12] Zhang Z, Wu L, Liang Y et al. J. Colloid Interface Sci., 1997, 188: 501.
[13] Lu X, Zhang Z, Liang Y. Langmuir, 1996, 12: 5501.
[14] Liang Y, Lu X, Li C. Acta Chim. Sin., 2000, 58: 742.
[15] Lu X, Zhang Z, Liang Y. J. Chem. Soc., Chem. Commun., 1994, 2731.
[16] Lu X, Zhang Z, Liang Y. Langmuir, 1997, 13: 538.
[17] Li C, Lu X, Luo X et al. Chem. Commun., 2001, 1440.
[18] Li C, Lu X, Liang Y. Langmuir, 2002, 18: 575.
[19] Ghosh P, Sengupta S, Bharadwaj P K et al. Langmuir, 1998, 14: 5712.
[20] Luo X, Wu S, Liang Y. Chem. Commun., 2002, 492.
[21] Luo X, Miao W, Wu S et al. Langmuir, 2002, 18: 9611.
[22] Lin Y, Kachar B, Welss, R G. J. Am. Chem. Soc., 1989, 111: 5542.
[23] Bhattacharya S, Krishnan-Ghosh Y. Chem. Commun., 2001, 185.
[24] Luo X, Liu B, Liang Y. Chem. Commun., 2001, 1556.
[25] Luo X, Li C, Liang Y. Chem. Commun., 2000, 2091.
[26] Kunitake T, Okahata Y. J. Am. Chem. Soc., 1979, 101: 5231.
[27] Wang X, Shen Y, Pan Y et al. Langmuir, 2001, 17: 3162.
[28] Wang X, Liang Y. J. Colloid Interface Sci., 2001, 233: 364.
[29] Wang X, Shen Y, Pan Y et al. Langmuir, 2000, 16: 7538.
[30] Luo X, Wang X, Wu S et al. J. Colloid Interface Sci., 2003, 258: 432.
[31] Nagel J, Oertel U, Friedel P et al. Langmuir, 1997, 13: 4693.
[32] Pang S, Huang J, Liang Y. Spectrosc. Lett., 2000, 33: 301.
[33] Pang S, Li C, Huang J et al. Colloid Surfaces A, 2001, 178: 143.
[34] Pang S, Li C, Huang J et al. Mater. Sci. Eng. C, 2000, 11: 137.
[35] Pang S, Liang Y. Mater. Chem. Phys., 2001, 71: 103.
[36] Pang S, Liang Y. Spectrochim. Acta, 2001, 57A: 435.
[37] Pang S, Liang Y. J. Colloid Interface Sci., 2000, 231: 59.
[38] Bohmer V. Angew. Chem. Int. Ed. Engl., 1995, 34: 713.
[39] Pang S, Ye Z, Li C et al. J. Colloid Interface Sci., 2001, 240: 480.
[40] Kraft A, Grimsdade A C, Holmes A B. Angew. Chem. Int. Ed. Engl., 1998, 37: 403.
[41] Wu Z, Wu S, Liang Y. Langmuir, 2001, 17: 7276.
[42] Wu Z, Wu S, Liao J H et al. Synth. Met., 2002, 130: 35.
[43] Wu Z, Wu S, Liang Y. Spectrochim. Acta, 2003, 59A: 1631.
[44] Roncali J. Chem. Rev., 1997, 97: 173.
[45] Wu Z, Wu S, Lu Z et al. J. Colloid Interface Sci., 2002, 251: 125.
[46] Chen W C, Jenekhe S A. Marcomolecules, 1995, 28: 454.
[47] Stine K J, Uang J Y-J, Dingnam S D. Langmuir, 1993, 9: 2112.
[48] Parazak D P, Uang J Y-J, Turner B et al. Langmuir, 1994, 10: 3787.
[49] Gericke A, Huhnerfuss H. Langmuir, 1994, 10: 3782.
[50] Huhnerfuss H, Neumann V, Stine K J. Langmuir, 1996, 12: 2561.
[51] Hoffmann F, Huhnerfuss H, Stine K J. Langmuir, 1998, 14: 4525.
[52] Du X, Shi B, Liang Y. Langmuir, 1998, 14: 3631.
[53] Du X, Shi B, Liang Y. Spectrosc. Lett., 1999, 32: 1.
[54] Du X, Liang Y. Chem. Phys. Lett., 1999, 313: 565.
[55] Du X, Liang Y. J. Phys. Chem. B, 2000, 104: 10047.
[56] Du X, Liang Y. Langmuir, 2000, 16: 3422.
[57] Du X, Liang Y. J. Phys. Chem. B, 2001, 105: 6092.
[58] Zhang Z, Liang Y, Tian Y et al. Spectrosc. Lett., 1996, 29: 321.
[59] Tieke B. Adv. Mater., 1990, 2: 222.
[60] Marshbanks T L, Ahn D J, Franses E I. Langmuir, 1994, 10: 276.
[61] Marshbanks T L, Franses E I. J. Phys. Chem. 1994, 98: 2116.
[62] Li C, Zhao B, Lu Y et al. J. Colloid Interface Sci., 2001, 235: 59.
[63] Ariga K, Kunitake T. Acc. Chem. Res., 1998, 31: 371.
[64] Kunitake T. Supramol. Sci., 1996, 3: 45.
[65] Moy V, Florn E, Gaub H. Science, 1994, 266: 257.
[66] Bohanon T, Denzinger S, Fink R. Angew. Chem. Int. Ed. Engl., 1993, 32: 1033.
[67] Huang J, Ding D, Zhang Z et al. Synth. Commun., 1998, 27: 681.
[68] Beukers R, Berends W. Biochim. Biophys. Acta, 1960, 41: 550.
[69] Ding D, Zhang Z, Shi B et al. Colloids Surf. A, 1996, 12: 25.
[70] Ahlers M, Ringsdorf H, Rosemeyer H et al. Colloid Polym. Sci., 1990, 268: 132.
[71] Li C, Huang J, Liang Y. Spectrochim. Acta, 2001, 57A: 1587.
[72] Huang J, Liang Y. Spectrosc. Lett., 1997, 30: 1441.
[73] Huang J, Liang Y. Thin Solid Films, 1998, 326: 217.
[74] Huang J, Liang Y. Thin Solid Films, 1998, 325: 210.
[75] Li C, Huang J, Liang Y. Langmuir, 2000, 16: 7701.
[76] Li C, Huang J, Liang Y. Langmuir, 2001, 17: 2228.
[77] Huang J, Li C, Liang Y. Langmuir, 2000, 16: 3937.
[78] Miao W, Du X, Liang Y. Langmuir, 2003, 19: 5389.
[79] Miao W, Luo X, Liang Y. Spectrochim. Acta, 2003, 59A: 1045.
[80] Pinnavaia T J, Todd Milws H, Becker E D. J. Am. Chem. Soc., 1975, 97: 7198.
[81] Green J J, Ying L, Klenerman D et al. J. Am. Chem. Soc., 2003, 125: 3763.
[82] Miao W, Luo X, Wu S et al. Spectrochim. Acta, Part A, in press.
[83] Miao W. Ph. D Thesis, Nanjing University, 2003.