http://www.chemistrymag.org/cji/2004/066043pe.htm |
Jun. 1,
2004 Vol.6 No.6 P.43 Copyright |
(State Key Laboratory of Functional Polymer Materials for Adsorption and Separation, Institute of Polymer Chemistry, Nankai University, Tianjin 300071, China)
Received Feb. 27, 2004; Supported by the National Natural Science Foundation of China (20074017)
Abstract Polystyrene N-hydroxyl
sulfonamide resin 1 and its derivative N-hydroxyl sulfonamide resins 2-5
were prepared and their corresponding N,O-diacetyl sulfonhydroxamate resins were used to
synthesize the ester with n-butanol and acetic anhydride. The mechanism of
esterification was that: O-H and N-H of N-hydroxyl sulfonamide resin reacted with acetic
anhydride respectively to form the active intermediate polystyrene N,O-diacetyl
sulfonhydroxamate, which was cleaved by n-butanol to produce n-butyl
acetate. The characterization of the specificity of resins 1-5 was determined by
the rate constant k1 calculated according to a two-substrate reversible
kinetic model. The results revealed that the activity of alkyl N-hydroxyl sulfonamide
resin 4 and resin 5 with the rate constant k1 of
3.74��10-3 M-1 min-1 and 9.13��10-3 M-1
min-1 were higher than those of the aryl N-hydroxyl sulfonamide resin 1
and resin 3. Resin 2 prepared from recycling resin 1 for 7 times
showed a higher activity than resin 1. Resin 3 with the benzene ring
substituted by the electron donating group gave the lowest activity with the k1
of 0.75��10-3 M-1 min-1.
Keywords polystyrene N-hydroxyl sulfonamide; N,O-diacetyl sulfonhydroxamate;
esterification; double acyl transfer
In recent years, polymeric reagents, containing a reactive functional group attached to a macromolecular backbone, have been utilized in many chemical reactions [1, 2]. The polymeric reagents could share both chemical and physical properties of the functional group and the polymeric matrix. Polymeric backbone could provide a specific microenvironment and played a decisive role in indicating the nature and reactivity of the attached functional group [3]. It could influence significantly on the selection of reaction solvent and the extent of functional group conversions in the subsequent reactions [4]. Introducing a new functional group to the polymeric matrix generally led to a variety of reactivity, selectivity [5-7]. A number of polymeric reagents have been prepared since the invention of Merrifield resin. Among them, the acylating reagents, such as polymer-bound nitrophenol [8, 9], HOBT (1-hydroxybenzotriazole) [10], hydroxamic acid [11], oximino esters [12], N-hydroxysuccinimide [13], or oximino dithiocarbonic anhydride [14] were especially important for their wide application in the acylation of alcohols, amines, and in the synthesis of peptides.
In this paper a new polymeric reagent -- polystyrene N-hydroxyl sulfonamide resin 1 and its derivative resins 2-5 were prepared and the applications of their corresponding N,O-diacetyl sulfonhydroxamate in the preparation of ester from n-butanol and acetic anhydride were investigated. The mechanism of esterification by resin 1 was studied to be a double acyl process. Characterization of the specificity of resins 1- 5 was discussed by a kinetic method.

Scheme 1 Resins 1-5 2 EXPERIMENTAL
2.1 General
Infrared spectra were recorded on a Bio-Rad FTS 13t spectrometer with KBr pellets. The CHN elementary analysis was recorded on Fess Heraes instrument and the Cl content of the resin was determined by the Volhard method. All the reagents were purified and dried following the literature. Polystyryl sulfonyl chloride resin (2.50 mmol/g, 1%DVB crosslinked, 100-200 mesh) and polystyryl chloromethyl resin (1.24 mmol/g, 1%DVB crosslinked, 100-200 mesh) were provided by Tianjin Nankai Hecheng Co., Tianjin, China.
2.2 Preparation of polystyrene N-hydroxyl sulfonamide resin 1
To polystyrene sulfonyl chloride resin (10 g, 25.2 mmol) in pyridine (200 mL) was added hydroxylamine hydrochloride (13.2 g, 0.252 mol). The mixture was stirred at room temperature for 8 h. Then the resin was collected by filtration, washed sequentially with DMF, DCM, Et2O and dried in a vacuum oven for 24 h. The loading was 2.52 mmol/g by CHN elementary analysis.
2.3 Preparation of polystyrene N,O-diacetyl sulfonhydroxamate resin 2
Resin 1 was used to prepare n-butyl acetate from n-butanol and acetic anhydride at 50��0.5ºC for 4 h according to the reaction process mentioned in 2.7 section. After repeating this process for 7 times, resin 2 was obtained with the N content of 2.89%.
2.4 Preparation of 4-(polystyryl benzyloxyl) benzene N-hydroxyl sulfonamide resin 3
4-Hydroxybenzene sulfonate sodium salt was obtained by treating 4-hydroxybenzene sulfonic acid with NaOH. Sodium methoxide (1.0 g, 18.5 mmol) was added to a suspension of chloromethylated polystyrene resin (3.0 g, 3.7 mmol) and anhydrous 4-hydroxybenzene sulfonate sodium salt (3.6 g, 18.5 mmol) in N, N-dimethyl acetamide (DMAc, 50 mL) [15]. The mixture was stirred at 90ºC for 2 days. The resulted resin was collected by filtration, washed sequentially by DMAc, H2O. The resin was acdified by 1 M HCl, then washed by H2O, MeOH, DCM, Et2O, and dried in vacuum oven at 25ºC for 24 h. The residual Cl content of the sulfonic acid resin was determined to be 0.6%. Pyridine (5 mL) was added to the sulfonic acid resin in DMF (10 mL) and the mixture was stirred at room temperature for 2 h. The resin was filtered, washed 3 times with DMF. Then the resin was suspended in DMF(15mL) and treated with thionyl chloride (5 mL) at 0ºC for 1 h, and the reaction was continued at room temperature for 20 h. The resin was filtered, washed with DMF, DCM, Et2O and dried in vacuum. The sulfonyl chloride resin was converted to N-hydroxyl sulfonamide resin 3 by reacting with hydroxylamine hydrochloride at 50 ºC for 20 h. The elementary analysis showed the N content of resin 3 was 1.10 mmol/g.
2.5 Preparation of 2-(Polystyryl sulfonyloxyl) ethyl N-hydroxyl sulfonamide resin 4
2-Hydroxyl ethyl sulfonate sodium salt was prepared by refluxing 2-chloroethanol with sodium sulfite for 4 h [16]. To a suspension of polystyryl sulfonyl chloride resin (0.50g, 1.3 mmol) and 2-hydroxyl ethyl sulfonate sodium salt (2.2 g, 12.6 mmol) in DMF (30 mL) were added pyridine (0.36 mL) and deionized water (36 mL) and the mixture was stirred at 90ºC for 2 days. The resin was filtered, washed by H2O, DMF, DCM, Et2O and dried in a vacuum oven at 25ºC for 24 h to afford (2-polystyryl sulfonyloxyl) ethyl sulfonate sodium salt. The residual Cl of the resin was determined to be 0.32%. The resin was acidified by 1 M HCl, and then converted to the corresponding N-hydroxyl sulfonamide resin 4 in a similar way as resin 3. The N content of the resin 4 was 2.18 mmol/g.
2.6 Preparation of 6-(polystyryl sulfonyloxyl) hexyl N-sulfonamide resin 5
The resin 5 was prepared in a similar way as resin 4 with the N content of 1.92 mmol/g.
2.7 General procedure of esterification
The esterification of n-butanol (200 eq.) and acetic anhydride (200 eq.) was performed in a reactor containing polystyrene N-hydroxyl sulfonamide resin (1 eq.) at 50��0.5ºC for 7 h. A 100 ml of reaction solution was taken out from the system every 30 min interval. Esterification conversion was calculated by first adding enough standard sodium hydroxide aqueous solution into the 100 ml reaction mixture, then titrating the excess base by the standard HCl aqueous solution.
2.8 Kinetic model of a two-substrate reversible reaction mechanism [17]
The kinetic model based on the two-substrate reversible reaction was elaborated by Han et al [18].

Where SA and SB denote the substrates (SA being the limiting substrate), PP and PQ the products, and k1 and k-1 are the rate constants of the direct and reverse process.
The rate PP expression according to this model can be represented by Eq. (1):
Where A and B are the concentrations of the substrates, P and Q are the concentrations of the products at reaction time t.
The integrated rate equation corresponding to this mechanism results in the parametric Eq. (2), containing two parameters (XE and e) that can be determined by optimization:
where A0 and B0 are the initial concentrations of the two substrates, X is the fractional conversion of the limiting substrate [X = (A0-A) / A0] , XE the equilibrium conversion, t the reaction time, and �� a parameter related to the rate constant k1, as the results from Eq. (2). The rate constant of the direct reaction can be calculated using the following equation:
Optimization by using Origin 6.0 gives the values of parameters XE and e that ensure the best fit of the model curve X = f(t) resulted from Eq. (2) with the experimental data points.
Eq. (2) represents a kinetic model for reversible two-substrate reactions by resins 1-5, which does not take account of the consecutive intermediate. Consequently, it is not accurate to describe the intimate reaction mechanism, but could be useful for specificity determination. The kinetic constant k1 is appropriate to be considered a measure of resin specificity under given reaction conditions, requiring one single measurement for each substrate.
3. RESULTS AND DISCUSSION
3. 1 Esterification by resins 1-5 and the acyl transfer mechanism
Resins 1-5 were prepared in our experiment. Resin 1 is the polystyrene N-hydroxyl sulfonamide resin. Resin 2 is the polystyrene N,O-diacetyl sulfonhydroxamate resin by recycling resin 1 for 7 times to prepare the n-butyl acetate from n-butanol and acetic anhydride. Resin 3 is the 4-(polystyryl benzyloxyl) N-hydroxyl sulfonamide with the benzene ring substituted by an electron donating group. Resins 4 and 5 are the alkyl N-hydroxyl sulfonamide indirectly prepared through coupling the hydroxyl alkyl sulfonic acid sodium salt with the sulfonate ester linking to the polystyryl sulfonyl chloride resin. Though the sulfonate ester bond is not a good linker for its instability at basic reaction conditions, breakdown of the sulfonate ester was not observed by IR spectra in our experiment conditions.
In the following experiments, an example of esterification by resin 1 was chosen to study the reaction mechanism. The esterification of n-butanol and anhydride by resin 1 was carried out at 50��0.5ºC for 7 h. IR spectra of resin 1 was showed in Figure 1. It was interestingly found that infrared absorption at 1792 cm-1 first appeared at 75 min, corresponding to ester carbonyl group, then the infrared absorption appeared at 1716 cm-1, corresponding to amide carbonyl group. These were in consistent with the reported IR spectra of N,O-diacetyl benzene sulfonhydroxamate [19], indicating the formation of the active intermediate polystyrene N,O-diacetyl sulfonyhydroxamate. With the time going on, both intensity of the infrared absorption at 1792 cm-1 and 1716 cm-1 increased.
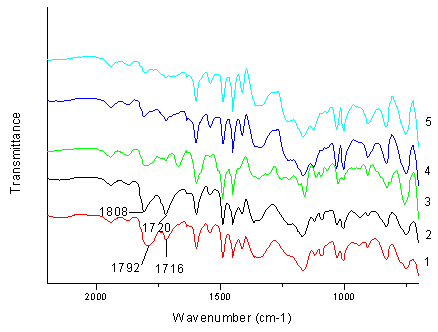
Fig. 1 IR spectra of the resins 1-5 after the esterification. Curve 1: Used resin 1. Curve 2: Resin 2. Curve 3: Used resin 3. Curve 4: Used resin 4. Curve 5: Used resin 5.

Scheme 2 Mechanism of the esterification of n-butanol and acetic anhydride by resin 1
Repeating the
esterification by resin 1 at 50ºC for 4 h, the conversion of n-butyl acetate
increased with the recycling times of the resin. After recycling the resin 1 for 7
times, the n-butyl acetate conversion reached a fixed value of 70%. The CHN
elementary analysis showed the N content of resin 1 used 7 times was 2.89%,
indicating that the N-H and O-H bonds of resin 1 were saturated by acetyl group. In
the other word, resin 1 was completely converted to polystyrene N,O-diacetyl
sulfonhydroxamate resin 2. Comparing with the resin 1 used only once, IR
spectra of resin 2 showed that the ester carbonyl absorption band shifted from 1792
cm-1 to 1808 cm-1 and the amide carbonyl absorption band changed
from 1716 cm-1 to 1720 cm-1, respectively (Figure 1).
Based on the experimental results above, the mechanism of
esterification by resin 1 was proposed as showed in Scheme 2: O-H bond of resin 1
first reacted with acetic anhydride to form the intermediate polystyrene O-acetyl
sulfonhydroxamate, then N-H was acetylated by acetic anhydride to form the more active
intermediate polystyrene N,O-diacetyl sulfonhydroxamate. Cleavage of the resin�Cbound O-acetyl sulfonhydroxamate and N,O-diacetyl
sulfonhydroxamate by n-butanol gave the target n-butyl acetate.
Furthermore, supposing that O-H and
N-H of resin 1 have the same reactivity and could be regarded as the same active
site during the esterification, the mechanism of esterification by resin 1
could be described by the following Scheme 3:

Scheme 3 The mechanism of esterfication by polystyrene
N-hydroxyl sulfonamide. E = the resin, AX = the acetic anhydride, A = acetic acid, EX =
the active intermediate polystyrene N, O-diacetyl sulfonhydroxamate, B = n-butanol,
BX= the product of amide.
According to this
mechanism, excess acetic anhydride AX (acyl donor) quickly react with the resin E to give
an acyl-resin intermediate EX, which then transfers the acyl group to the n-butanol
B (acyl acceptor).
3.2 Characterization of specificity of resins 1-5 in esterification
The esterifications by resins 2-5 were carried out at the same condition as resin 1.
In order to intuitively compare the characterization of the specificity of resins 1-5, a
kinetic method of a two-substrate reversible reaction mechanism was introduced and the
apparent rate constant k1 representing the whole reaction process was
calculated according to Eq. (2), Eq.(3).
A good agreement between the experimental data for resins 1-5
and the values of the substrate conversions calculated according to Eq.(2) was observed,
as shown in Table 1. Examples of esterification by resin 1, 4 were
shown in Figure 2.
Compared with the aryl N-hydroxyl sulfonamide resin 1 and resin 3,
the alkyl N-hydroxyl sulfonamide resin 4 and resin 5 have the higher
activity with the rate constant k1 of 3.74��10-3 M-1
min-1 and 9.13��10-3 M-1 min-1, respectively.
From the IR spectra of the used resin 4 and 5 (Fig. 2), the active
intermediate contents were observed to be very low. This demonstrated that the
alkyl-intermediate was more active and easier to be cleaved by n-butanol to form
the n-butyl acetate, and led to the higher rate constant k1. The
activity of resin 2 (k1 = 3.88��10-3 M-1
min-1) is 4 fold more over that of resin 1 (k1 = 0.80��10-3
M-1 min-1), which could be interpreted by the IR spectra in
Figure 2. The stronger absorption of the ester and amide carbonyl group in resin 2
demonstrated that resin 2 contained the saturated content of the active
intermediate and had the higher activity of esterification. The 4-methoxyl substituted
N-hydroxyl benzene sulfonamide (resin 3) behaved lower activity ( k1
= 0.75��10-3 M-1 min-1, lower than that of resin 1). It
might be explained by that the electron-donating group on benzene ring of resin 3 played a
role of stabilization of N-hydroxyl sulfonamide and made it more difficult to react with
acetic anhydride, causing the low intermediate content.


Fig. 2 Kinetic model of esterification by resin 1
and resin 4. The points are the average of three experimental data and the lines
represent the values calculated according to Eq.(2)
��
��
Table 1 Calculated kinetic constants for the esterification by resins 1-5 according to Eq.(2) and Eq.(3)
Optimized parameters |
Kinetic rate constant |
||
XE (P1) |
e (P2) |
||
Resin 1 |
0.740 |
-0.00302 |
0.80 |
Resin 2 |
0.672 |
-0.0202 |
3.87 |
Resin 3 |
0.605 |
-0.00522 |
0.75 |
Resin 4 |
0.700 |
-0.0171 |
3.74 |
Resin 5 |
0.694 |
-0.0431 |
9.13 |
Polystyrene N-hydroxyl sulfonamide resin 1 and its derivative N-hydroxyl sulfonamide resin 2-5 were prepared and their corresponding N,O-diacetyl sulfonhydroxamate resins were used to synthesize the ester with n-butanol and acetic anhydride. During the esterification, polystyryl N-hydroxyl sulfonamide reacted with acetic anhydride to form the active intermediate polystyryl N,O-diacetyl sulfonhydroxamate, which was cleaved by n-butanol to form n-butyl acetate. Characterization of specificity of resins 1-5 was determined by a kinetic method. Alkyl N-hydroxyl sulfonamide resins 4 and 5 with the apparent rate k1 of 3.74��10-3 M-1 min-1 and 9.13��10-3 M-1 min-1, respectively, have the higher activity than the aryl N-hydroxyl sulfonamide 1 and resin 3. To the aryl N-hydroxyl sulfonamide, resin 3 with an electron donating substitution on the benzene ring showed the lower rate constant k1 of 0.75��10-3 M-1 min-1, compared with resin 1 of k1 = 0.80��10-3 M-1 min-1. Resin 2 prepared from recycling resin 1 for 7 times showed an increasing rate constant k1 of 3.87��10-3 M-1 min-1 value, which was 4 fold over that of resin 1. REFERENCES
[1] Fruchtel J J, Jung G. Angew. Chem. Int. Ed. Engl., 1996, 35: 17.
[2] Balkenhohl F, Bussche-Hunnefeld C, Lansky A, et al. Angew. Chem. Int. Ed. Engl., 1996, 35: 2288.
[3] Sreekumar K, Pillai V N R. Macromolecules, 1989, 22: 3303.
[4] Morawetz H. J. Polym. Sci. Symp., 1988, 72: 9.
[5] Georage B K, Pillai V N R. Polymer, 1989, 30: 179.
[6] Sudhakaran K V, Pillai V N R. Indian J. Chem., 1990, 29B: 1012.
[7] Devaky K S, Pillai V N R. Proc. Indian Acad. Sci. (Chem. Sci.), 1990, 102: 521.
[8] Cohen B J, Karoly-Hafeli H, Patchornik A. J. Org. Chem., 1984, 49: 922.
[9] Lee J W, Louie Y Q, Walsh D P, Chang Y T. J. Com. Chem., 2003, 5: 330.
[10] Dendrinos K, Jeong J, Huang W, Kalivretenos A G. Chem. Commun., 1998, 4: 499.
[11] Sophiamma P N, Sreekuman K. Eur. Polym. J., 1997, 33: 863.
[12] Kumari K A, Sceekumar K. Polymer, 1996, 37: 171.
[13] Chinchilla R, Dodsworth D J, Nájera C, Soriano J M. Tetrah. Lett., 2001, 42: 4487.
[14] Kumari K A, Sherlymole P B, Sreekumar K. Eur. Polym. J., 1995, 31: 565.
[15] Zhong H-M, Greco M N, Maryanoff B E. J. Org. Chem., 1997, 62: 9326.
[16] Klamann D, Bertsch H. Chem. Ber., 1955, 88: 204.
[17] Peter F, Preda G. J. Mol. Cataly. B: Enzyme, 2002, 19-20: 467.
[18] Han D J S, Lee S B et al. Biotechnol. Bioeng., 1987, 30: 239.
[19] Smith P A S, Hein G E. J. Am. Chem. Soc., 1960, 82: 5731.