http://www.chemistrymag.org/cji/2004/067047pc.htm |
Jul. 1,
2004 Vol.6 No.7 P.47 Copyright |
Shen Haoyu1,2 Liao Daizheng3
(1 Ningbo Academy of Agriculture Sciences, 6th bridge Ningchuan
Road, Ningbo Zhejiang, PR China 315040; 2 Institute of Chemical &
Engineering Sciences, Singapore, 139959, 3 Chemistry Department, Nankai
University,Tianjin, 300071)
Abstract The oxidation of alcohols
into corresponding aldehydes and ketones is a crucial transformation in organic chemistry,
with both of academic and industry relevance. Recently, more and more efforts
have been put in highly valuable catalytic oxidizing process. Among these, protocols based
on O2, air or H2O2 are particularly attractive because of
cheap and readily available oxidants, with H2O as the only byproduct. Succeed
examples include homogeneous catalysts (e.g. Ru-, Pd-, Cu- complexes; bi-metallic
complexes systems) and heterogeneous catalysts (e.g. metal catalysts and supported
catalysts, including mesoporous materials, zeolites, etc.). In this paper, the
synthesis, characterization, and catalytic studies of four ruthenium-based homogeneous
catalysts (Ru(bpy)2(OX).4H2O (1), Ru(4,4’dmbpy)2 (OX).4H2O (2),Ru(phen)2(OX).4H2O (3) and Ru(5-NO2phen)2(OX).4H2O
(4)) have been reported for this purpose. Some effects, such as of solvent,
temperature, reaction time etc on these catalytic reactions, have been studied. The
results showed that the four catalysts could be used for selective oxidation of
alcohol to corresponding aldehydes or katones. The conversion of alcohol is higher at
higher temperature (80 oC) than at room temperature (25 oC). Solvent
effects on these catalytic reactions have not been observed obviously. The catalytic
reactions are much faster when electron-pushing ligands contained catalyst is used, vice
versa. Presumed mechanism has been presented. It is found that oxalate ligand plays an
important role in the catalytic process. All the four catalysts can be recycled and reused
for at least 10 runs, with the conversion of alcohol higher than 80%.
Keywords Selective Oxidation of Alcohols, Homogeneous Ru-catalysts, Synthesis,
Characterization, Catalytic properties.
均相钌催化剂的合成、表征及其在醇的选择性氧化中的催化性能研究
沈昊宇1,2* 廖代正3(1 宁波市农业科学研究院 浙江宁波 315040; 2 Institute of Chemical & Engineering Sciences, Singapore,139959 3南开大学化学系 天津 310071)
2004年4月5日收稿
摘要 报道了4种均相钌催化剂(Ru(bpy)2(OX)
.4H2O (1), Ru(4,4'dmbpy)2(OX).4H2O (2),Ru(phen)2(OX).4H2O
(3)和 Ru(5-NO2phen)2(OX).4H2O (4))的设计与合成,并研究了它们在醇的选择性催化氧化中的性质。通过考察溶剂、反应温度等因素优化了某些催化剂的使用条件。研究了配体和功能团调节对于催化剂的微结构的影响及构效相关性等。结果表明,这四种催化剂对芳香醇和脂肪醇均可以有效地选择性催化氧化为相应地醛或酮。醇的转化率在反应温度较高时(80
oC)较室温(25 oC)时略高。采用不同溶剂对于醇的转化率和产物的选择性均无明显的影响。当催化剂含有推电子基团的配体时,反应可以更快速地进行;而当催化剂含有拉电子基团的配体时,反应则相对进行缓慢。提出了催化剂可能的催化机理,结果发现草酸根配体在催化过程中起到十分关键的作用。对催化剂的回收率与循环使用的实验研究发现,催化剂循环使用10次后,醇的转化率仍可达到80%以上。催化剂可以回收再利用。
关键词
醇的选择性催化氧化;均相钌催化剂;合成;表征;催化性质
将醇选择性的氧化成为醛或酮是有机化学中一重要的反应,具有重要的理论研究价值和实用价值[1]。传统方法中,通常采用无机氧化物,如Cr(VI)试剂来实现这一类反应。然而,这类试剂需要计量级投入到反应中;而且通常是有毒有害的,很难从产物中分离;因此是环境不友好也不经济的。更重要的是,大部分这类试剂都难实现多种醇的选择性氧化,特别是那些含有易氧化基团(如不饱和基团或杂原子等)的醇的选择性氧化[2]。近年来,越来越多的科学家致力于高附加值催化氧化过程的研究。其中,采用O2, 空气或H2O2 [3]为氧化剂的研究尤为热烈;这主要是由于这些氧化剂便宜、易得且环境友好。成功的实例包括:Ru-, Pd-, Cu- 及双金属化合物均相催化剂[4-12]和金属催化剂、以中孔材料,沸石等为载体的异相催化剂等[5-20]。本文报道了4种均相钌催化剂的设计与合成,并研究了它们在醇的选择性催化氧化中的性质。通过考察溶剂、反应温度等因素优化了某些催化剂的使用条件。研究了配体和功能团调节对于催化剂的微结构的影响及构效相关性等。本文还就可能的催化机理和催化剂的回收与循环使用进行了研究和讨论。
1 实验部分
1.1 主要仪器和试剂
EURO EA elemental analyzer CAP 40/2型元素分析仪; ICP-OES 型等离子发射光谱仪;Bio-Rad EXCAL3000型红外光谱仪; Shimadzu UV-2550
型紫外---可见分光光度计; Auto Lab -100电化学分析仪;TA instrument SDT 2960 simultaneous DSC-TGA型热分析仪;Agilent HP-6890N气相色谱;Agilent HP-6890N/MS-5793气相色谱----质谱联用仪;Bruker 400 MHz核磁共振仪;所用试剂均为分析纯试剂。
1.2 催化剂的合成
催化剂Ru(bpy)2(OX) .4H2O (1)(bpy为2,2'-联吡啶,OX为草酸根)是Fe(bpy)3(ClO4)2(ClO4为高氯酸根)与K3Ru(OX)3通过氧化还原和配体取代反应得到:在Fe(bpy)3(ClO4)2 (0.5 mmol 364.4 mg) 的5 mL水溶液中,搅拌下滴加K3Ru(OX)3
(0.5 mmol, 241 mg)的5 mL水溶液。滤去反应后的不溶物,得深红色溶液,室温下静置约3个月,得到深红色晶体,过滤,晶体用少量水和乙醇洗涤,真空干燥,产率32%。催化剂Ru(4,4’dmbpy)2(OX).4H2O
(2) (4,4'dmbpy为4,4'-二甲基2,2'-联吡啶),Ru(phen)2(OX).4H2O
(3) (phen 为邻啡罗啉)和
Ru(5-NO2phen)2(OX).4H2O (4) (5-NO2phen 为5-硝基邻啡罗啉)的合成方法与(1)类似,分别使用Fe(4,4'dmbpy)3 (ClO4)2,Fe(phen)3(ClO4)2和Fe(5-NO2phen)3(ClO4)2与K3Ru(OX)3反应,得到深红色和褐色微晶。
1.3 催化剂的催化活性研究实验
以Ru(bpy)2(OX) .4H2O为催化剂,选择性催化氧化肉桂醇为例。5g肉桂醇溶于10mL正己烷中,升温至80 oC,搅拌回流下缓慢加入15mg(0.3 wt%)的催化剂Ru(bpy)2(OX) .4H2O,1 mL H2O2。每隔15分钟取样,过硅胶小柱,正己烷洗脱,产物以GC-MS和NMR定性,GC定量分析。采用其他催化剂或底物的实验方法与之类似。醇的转化率和产物选择性的计算公式分别为式(1)和(2):
转化率(%)= [(A0-At)/A0]´ 100% (1)
其中,At:时间为t时的反应物的GC峰面积
A0:时间为t0时的初始反应物的GC峰面积
选择性(%)= [npi /å npi]´ 100% (2)
其中,npi:时间为t时的目标产物的量
å npi:时间为t时的所有产物的量
1.4 催化剂的回收与循环利用实验
催化剂的回收:以Ru(bpy)2(OX).4H2O为例,反应后的溶液减压浓缩至近干,乙醚中重结晶,红外光谱定性表征回收后的催化剂。催化剂循环利用的实验方法与1.3所述的催化剂的催化活性研究实验类似。
2. 结果与讨论
2.1催化剂Ru(bpy)2(OX)
.4H2O (1)的晶体结构
催化剂(1)的分子结构图见图1, Ru(II)与两个2,2’-联吡啶的4个氮原子和草酸根的其中两个氧原子配位[Ru-N(4) =
2.0181(8)Å, Ru-N(3) = 2.039(7) Å, Ru-N(2) = 2.026(7) Å, Ru-N(1) =
2.044(7) Å, Ru-O(2) = 2.044(7) Å, Ru-O(1) = 2.087(7) Å] Ru(II)离子处于六配位的畸变八面体的配位环境中。Ru(II)的配位单元进一步由氢键相联而构成三维骨架。如图2所示,在该配合物的晶胞图中,OX2-中的配位氧原子O2和未配位的氧原子(O4, O3)与水分子OW3, OW4和OW2通过氢键O2…H-OW3 = 2.815 Å, O4…H-OW4 =
2.784 Å, O3…H-OW2 = 2.689 Å相联;而OW3和OW4除通过氢键OW3-H…OW4 = 2.731 Å相互联结外,还进一步分别与OW1和OW2以氢键(OW3-H…OW1 = 2.719 Å, OW4-H…OW2 = 2.756 Å)相联而形成一个八员环。同时,相邻的OW2和OW1之间也彼此通过氢键OW1…H-OW2 = 2.824 Å在空间形成一个四员环。两个环之间的二面角为6.5° 。由此而形成氢键联结的四员环和八员环交替的三维结构。

图1 催化剂Ru(bpy)2(OX) .4H2O
(1)的分子结构图

图2 催化剂Ru(bpy)2(OX) .4H2O
(1)的晶胞图
2
.2催化剂组成及物理性质催化剂(1)-(4)的元素分析实验值和理论计算值(各元素的质量百分比)列于表1;一般物理性质列于表2,以DMF为溶剂测定了配合物的电导,数据表明配合物均为非电解质。
表1催化剂(1)-(4)的元素分析结果
编号 |
化学式 |
元素分析值( %)实验值(理论值) |
|||
C |
H |
N |
Ru |
||
1 |
RuC22H28N4O8 |
46.25(45.97) |
4.58(4.89) |
9.62(9.70) |
17.30(17.50) |
2 |
RuC26H38N4O8 |
48.98(49.13) |
6.28(6.03) |
8.55(8.81) |
15.68(15.90) |
3 |
RuC26H32N4O8 |
49.42(49.60) |
5.55(5.12) |
8.54(8.90) |
16.28(16.05) |
4 |
RuC26H30N6O12 |
43.42(43.40) |
4.55(4.20) |
11.54(11.68) |
14.28(14.04) |
表
2 催化剂(1)-(4)的物理性质编号 |
催化剂 |
产率 % |
熔点 ℃ |
颜色状态 |
电导 |
1 |
Ru(bpy)2(OX) .4H2O |
32 |
>280 |
深红色晶体 |
0.68 |
2 |
Ru(4,4’dmbpy)2(OX).4H2O |
63 |
>280 |
深红色微晶 |
0.69 |
3 |
Ru(phen)2(OX).4H2O |
58 |
>280 |
深红色微晶 |
0.85 |
4 |
Ru(5- NO2phen)2(OX).4H2O |
45 |
>280 |
褐色微晶 |
0.56 |
2
.3 催化剂的光谱和电化学性质2.3.1 红外光谱
上述四种催化剂在红外光谱上均存在特征吸收峰: 1640, 1610 cm-1的强峰可归属为草酸根的n (C=O)伸缩振动吸收; 1480, 1420和850cm-1的强吸收峰则归属于配体bpy,4,4’dmbpy,phen和5-NO2phen的 C=C 和 C=N 的振动吸收。
2.3.2 ESR
上述四种催化剂的室温及110 K的ESR谱中没有检测到信号。这一现象表明催化剂中钌离子为二价且处于低自旋态。
2.3.3催化剂的电化学性质
测定了Fe(bpy)3(ClO4)2, K3Ru(OX)3和催化剂Ru(bpy)2(OX) .4H2O 的DMF溶液的循环伏安曲线。Ru(bpy)2(OX) .4H2O 仅在-1200 - -1000 mV出现了一对与原料K3Ru(OX)3相似但曲线走向相反的氧化还原峰,它们对应于RuII
2.4 催化剂的催化活性
2.4.1催化产物的选择性研究
采用GC-MS和NMR定性,GC定量考察了四种催化剂对不同底物,在不同实验条件下的催化反应。图3中给出了底物2-巯基乙醇以H2O2为氧化剂,Ru(bpy)2(OX).4H2O为催化剂时间t = 0和t = 2h时的GCMS总离子流图和质谱图。结果表明,在Ru(bpy)2(OX).4H2O催化作用下,2-巯基乙醇可以被选择性地氧化成为单一产物2-巯基乙醛。其他各催化剂均可以催化底物选择性地氧化成唯一的目标产物,即反应产物的选择性近100%,在以下的讨论中仅讨论各因素对反应的转化率的影响。
2.4.2催化剂Ru(bpy)2(OX) .4H2O (1)对8种醇的选择性催化氧化研究
以Ru(bpy)2(OX) .4H2O为催化剂研究了其在不同反应温度和溶剂中,以H2O2为氧化剂选择性催化氧化肉桂醇等8种底物,结果列于表3。结果表明,催化剂Ru(bpy)2 (OX) .4H2O对芳香醇和脂肪醇均可以有效地选择性催化氧化为相应地醛或酮。醇的转化率在反应温度较高时(80 oC)较室温(25 oC)时略高。采用不同溶剂(甲苯、水和二氯甲烷)对于醇的转化率和产物的选择性均无明显的影响。
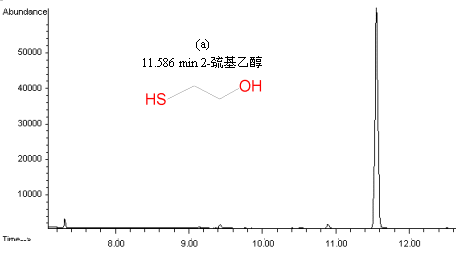
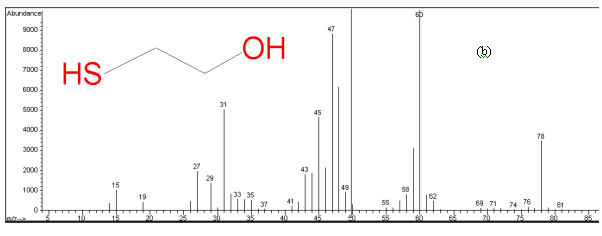

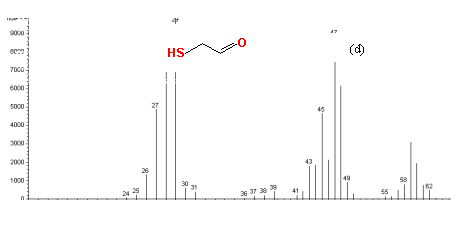
图
3底物2-巯基乙醇以H2O2为氧化剂,Ru(bpy)2(OX).4H2O为催化剂反应体系的GCMS总离子流图和质谱图 (a)时间t = 0时体系的GCMS总离子流图;(b)时间t = 0时保留时间为11.586 min的组分的质谱图(2-巯基乙醇);(c)时间t = 2h 时体系的GCMS总离子流图;(d)时间t = 2h 时保留时间为12.198 min的组分的质谱图(2-巯基乙醛)表
3 Ru(bpy)2(OX) .4H2O在不同反应温度和溶剂中醇的选择性催化氧化研究编号 |
底物 |
产物 |
转化率 (%) (80 oC/25 oC) |
||
甲苯 |
水 |
二氯甲烷 (a) |
|||
1 |
|
|
97/92 |
95/90 |
-/95 |
2 |
|
|
99/93 |
98/92 |
-/96 |
3 |
|
|
96/90 |
95/90 |
-/92 |
4 |
|
|
99/90 |
98/92 |
-/97 |
5 |
|
|
99/95 |
95/93 |
-/95 |
6 |
|
|
98/92 |
92/90 |
-/93 |
7 |
|
|
95/90 |
92/90 |
-/98 |
8 |
|
|
98/92 |
95/90 |
-/99 |




(a)采用CH2Cl2为溶剂时实验只在25 oC进行
2.4.3催化剂(1)-(4)对8种醇的选择性催化氧化研究比较
为了考察和比较不同催化剂,尤其是不同配体对于醇的选择性催化氧化影响,研究了催化剂(1)-(4)在80 oC,以甲苯为溶剂,H2O2为氧化剂对8种醇催化氧化研究,结果列于表4。其中以肉桂醇为底物,不同催化剂,不同反应时间转化率随时间的变化图如图4所示。结果表明,当催化剂含有推电子基团的配体时,反应可以更快速地进行;而当催化剂含有拉电子基团的配体时,反应则相对进行缓慢。如催化剂2较1配体中含有推电子基团,反应在1小时时,底物肉桂醇的转化率达97%,而同样的反应以1为催化剂则需2小时才可以达到相近的转化率(表4,1,图4)。
表4 催化剂(1)-(4)对醇的选择性催化氧化研究
编号 |
底物 |
产物 |
转化率 (%)/反应时间(h) |
|||
( 1) |
( 2) |
( 3) |
( 4) |
|||
1 |
|
|
97/2.0 |
96/1.0 |
95/4.0 |
94/18.0 |
2 |
|
|
99/2.0 |
98/1.0 |
95/4.0 |
93/18.0 |
3 |
|
|
96/2.0 |
96/1.0 |
92/4.0 |
91/18.0 |
4 |
|
|
99/2.0 |
98/1.0 |
95/4.0 |
94/18.0 |
5 |
|
|
99/2.0 |
98/1.0 |
92/4.0 |
92/18.0 |
6 |
|
|
98/2.0 |
98/1.0 |
94/4.0 |
93/18.0 |
7 |
|
|
95/2.0 |
95/1.0 |
93/4.0 |
92/18.0 |
8 |
|
|
98/2.0 |
97/1.0 |
92/4.0 |
92/18.0 |




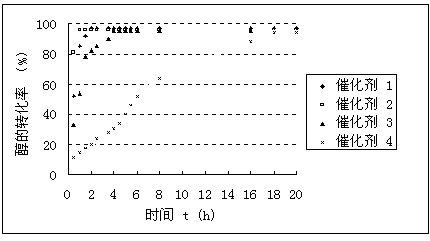
图
4催化剂(1)-(4)催化底物1的不同时间的转化率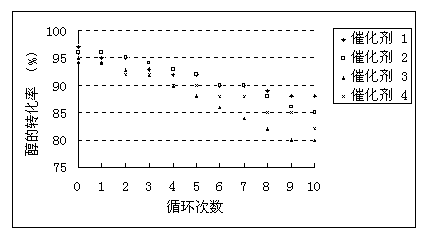
图5催化剂催化底物1的不同循环次数的转化率
2
.5 催化剂的回收与循环使用为了考察催化剂回收后的催化性能,研究了回收后的四种催化剂在80 oC,以甲苯为溶剂,H2O2为氧化剂催化氧化肉桂醇随循环次数的变化研究。各次循环后的结果见图5。结果表明,循环10次后各催化剂仍可以有效的催化肉桂醇选择性地氧化成肉桂醛。肉桂醇的转化率分别为催化剂(1) 88%(反应2 h),催化剂(2) 85%(反应1 h),催化剂(3) 80%(反应4 h),催化剂(4) 82%(反应18 h)。
2.6 可能的催化机理研究
为了研究催化剂的催化机理,考察了两种不含草酸根的Ru(II)化合物Ru(bpy)2Cl2和Ru(bpy)3Cl2在80 oC,以甲苯为溶剂,H2O2为氧化剂,肉桂醇为底物时的醇的氧化情况。结果发现,48 h后,肉桂醇的转化率几乎为零;由此推断催化剂中的草酸根在催化过程中起到十分关键的作用,这可能是由于它可以与H2O2形成氢键,构成稳定的8员环结构,有利于H2O2进一步进攻醇,而实现醇的选择性氧化。这一推断也与2.4.3中有关配体对于催化剂性能的影响的研究结果相符。即:催化剂含有推电子基团的配体时(电子向草酸根离域,更有利于与H2O2形成氢键),反应可以更快速地进行;而当催化剂含有拉电子基团的配体时,反应则相对进行缓慢。催化剂可能的催化机理如图6所示,过渡态的分离与表征研究正在进行中。
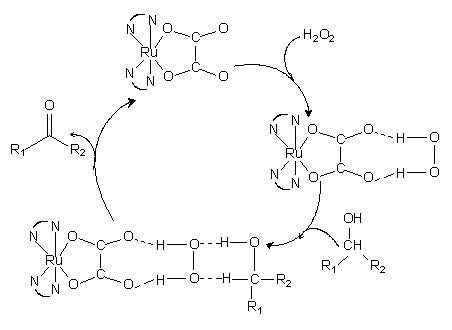
图6催化剂可能的催化机理
致谢 本研究工作得到了新加坡Agency for Science, Technology and Research (A*STAR) 的资助。本研究工作还得到了新加坡Institute of Chemical & Engineering Sciences的Dr. K. Carpenter, Prof. C. B. Ching 和Dr. Z. M. A. Judeh的支持与帮助。在表示感谢。
REFERENCES
[1] Larock R C. Comprehensive Organic Transformations. New York: VCH Publishers, 1989.
[2] Sheldon R A, Kochi J K. Metal-Catalyzed Oxidations of Organic Compounds. New York: Academic Press, 1981.
[3] Sato K, Aoki M, Noyori, R. Science, 1998, 281: 1646.
[4] Ligtenbarg A G, Hage J R, Fering B L. Coord. Chem. Rev., 2003, 237: 89.
[5] Besson M, Gallezot P. Catalysis Today, 2000, 57: 127.
[6] Sloboda-Rozner D, Alsters P L, Neumann R. J. Am. Chem. Soc., 2003, 125: 5280.
[7] Khenkin A M, Shimon L J W, Neumann R. Inorg. Chem., 2003, 42: 3331.
[8] Mahata N, Tanielyan A K, Augustine R L et al. Chemical Industries, 2003,89: 551.
[9] Poyatos M, Mata J A, Falomir E et al. Organometallics, 2003, 22: 1110.
[10] Blackburn L, Kanno H, Taylor R J K. Tetrahedron Letters, 2003, 44: 115.
[11] Surendra K, Krishnaveni N S, Reddy M A et al. J. Org. Chem., 2003, 68: 2058.
[12] Suzuki T, Morita K, Tsuchida M et al. J. Org. Chem., 2003, 68: 1601.
[13] Zhan B Z, White M A, Sham T K et al. J. Am. Chem. Soc. 2003, 125: 2195.
[14] Shaabani A, Teimouri F, Lee D G. Synth. Commun. 2003, 33: 1057.
[15] Bhar S, Chaudhuri S K. Tetrahedron 2003, 59: 3493.
[16] Griffin K G, Johnston P, Bennett S et al. Chemical Industries, 2003, 89: 169.
[17] Tanyeli C, Gumus A. Tetrahedron Letters, 2003, 44(8): 1639.
[18] Sakuratani K, Togo H. Synthesis, 2003, 1: 21.
[19] Biella S, Rossi M. Chemical Commun. 2003, 3: 378.
[20] Duliere E, Devillers M, Marchand-Brynaert J. Organometallics, 2003, 22: 804.