http://www.chemistrymag.org/cji/2004/069061ne.htm |
Sep. 1,
2004 Vol.6 No.9 P.61 Copyright |
Global geometry optimization of Na+(H2O)m clusters using a hybrid algorithm
Cao Yilin, Wang Yansong(College of Chemistry and Environmental Science, Henan Normal University, Xinxiang 453002)
Supported by the Natural Science Foundation of Henan Province (No.532221). Abstract A hybrid algorithm, based on genetic algorithm (GA), fast simulated annealing (FSA) and conjugated gradient algorithm (CGA) was proposed and used to optimize the structure of Na+(H2O)m (m=1-11) clusters. The empirical potential energy functions employed here included SPC, TIP, TIP3P, TIP4P and TIPS2. The optimized structure of Na+(H2O)m clusters by using these different potential energy functions were found to share the same configurations.
Key words Hybrid optimization algorithm, potential function, Na+(H2O)m clusters Many recent developments in the computational technology for random search have been made in the area of atomic and molecular cluster calculations. For example, Niesse used a modified GA method to optimize the geometry of atomic and molecular clusters[1]. Greguick proposed a modified deterministic/stochastic GA method for the determination of the global minimum of (Ar)n and B(Ar)n clusters. The deterministic / stochastic GA converged more quickly to the global minimum than other random search procedures[2]. As a global optimization algorithm, the simulated annealing algorithm has been widely used to optimize the molecular clusters too[3]. But, applying all these methods to larger systems is limited. To make the random search more efficient, based on GA, FSA and CGA we have presented a hybrid optimization algorithm and applied it to water clusters in the previous works. The results showed the structures of water clusters with lower potential energy were found and a more quick optimization strategy than using other was got. So, this method is proved more effective and it can be used to optimize the structure of water clusters in the electrostatic field of sodium ion in this paper. 1 POTENTIAL FUNCTION
The most stable structure of a cluster is usually the geometry with the lowest potential energy calculated through some potential functions. In the optimization of Na+(H2O) m clusters, the interaction energy between water molecule and sodium ion can be computed using the following equation[4]:
 (1)
Where the
index I denotes the sodium ion, the charge qI is +1, and the parameters AI,
BI are 447.69 kJ Å 6 /mol and 19.25 kJ Å 3 /mol, respectively. The interaction energy between
water molecules will be calculated according to equation 2:
(1)
Where the
index I denotes the sodium ion, the charge qI is +1, and the parameters AI,
BI are 447.69 kJ Å 6 /mol and 19.25 kJ Å 3 /mol, respectively. The interaction energy between
water molecules will be calculated according to equation 2:
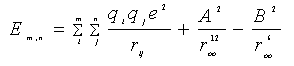 (2)
The
parameters of the potential functions are listed in Table 1. In this table, SPC, TIP,
TIP3P, TIP4P and TIPS2 are five kinds of potential energy functions which have the same
literal expression but with different parameters. The first 3 are three-site models, and
the last 2 are four-site models. The geometries of the models are shown in Figures 1. In
the four-site model, the negative charge is moved off the oxygen atom and towards the
hydrogen at a point (M) on the bisector of the HOH angle.
Table
1 Water monomer parameters for potential functions[5,6]
(2)
The
parameters of the potential functions are listed in Table 1. In this table, SPC, TIP,
TIP3P, TIP4P and TIPS2 are five kinds of potential energy functions which have the same
literal expression but with different parameters. The first 3 are three-site models, and
the last 2 are four-site models. The geometries of the models are shown in Figures 1. In
the four-site model, the negative charge is moved off the oxygen atom and towards the
hydrogen at a point (M) on the bisector of the HOH angle.
Table
1 Water monomer parameters for potential functions[5,6]
SPC |
TIP |
TIP3P |
TIP4P |
TIPS2 |
|
R(OH), Å |
1.0000 |
0.9572 |
0.9572 |
0.9572 |
0.9572 |
∠HOH ,deg |
109.47 |
104.52 |
104.52 |
104.52 |
104.52 |
A2*10-3,kJ Å12/mol |
2633.41 |
2426.72 |
2435.09 |
2510.40 |
2907.88 |
B2, kJ Å 6/mol |
2617.09 |
2196.6 |
2489.48 |
2552.24 |
2510.40 |
q(O) |
-0.820 |
-0.800 |
-0.834 |
||
q(H) |
0.41 |
0.40 |
0.417 |
0.52 |
0.535 |
q(M) |
1.04 |
1.07 |
|||
r(OM), Å |
0.15 |
0.15 |

(a) (b)
Figure 1 3-site (a) and 4-site (b) water models
2 METHOD
In this paper, the water molecule was treated as a rigid molecule and could be completely described by six coordinates (Rx, Ry, Rz, q, F, f). The molecule's initial coordinate (Rx, Ry, Rz) were generated randomly within a cubic box of size L3 centered at the origin. We took R = L (d-0.5), where L = 4.0 *(6*n)1/3, n is the number of water molecules and ion3. d is a random number between 0 and 1. q, F and f are generated randomly on the interval [0, 2p]. In addition, we put the sodium ion at the center of the box with the coordinate of (0.0, 0.0, 0.0, 0.0, 0.0, 0.0). In our procedure, 30 populations with 30 individuals each were produced. Then the best individual in each population was selected to form the initial population. In our hybrid optimization algorithm, the basic structure of GA was used, but the search strategy was improved. With regard to crossover, geometric mean, arithmetic mean, two-point crossover, one-point crossover and n-point crossover were employed; as regard to mutation, inversion and FSA operators were involved. In every generation, the crossover and mutation were performed first to give a coarse solution to the problem, and then the CGA was performed to have local search for the better solution. The process was performed iteratively. The program was terminated while the change in the potential energy was smaller than 0.04 kJ/mol in 500 generations. 3 RESULTS AND DISCUSSION
We employed five kinds of water potential functions to optimize the structure of Na+(H2O)m clusters. The results show: the geometries of Na+(H2O)m clusters for the five potential functions share the same configurations, have a little difference in the distance of Na+ and H2O which is so little that can be ignored. Here, only the geometry structures of Na+(H2O)m clusters which were optimized by using TIPS2 are given. In Figure 2, the line between sodium ion and oxygen atom in water molecules does not stand for a chemical bond, it is used to help understanding the geometry of the clusters. The dotted line between water molecules represents a hydrogen bond. At the beginning, the water molecules arrange around the Na+ symmetrically and form the first shell or the inner wedge into the first shell. With the increasing in m, instead they will be distributing around the first shell of forming hydrogen bond with the water molecules in the first shell. The extrusion of water molecules may be caused by the repulsion interactions between water molecules in the first shell and the formation of hydrogen bond is favourable for decreasing the energy of the clusters while the fifth and the fallowing water molecules are added to the clusters. As a result, the water molecules gradually filled into the second shell and aggregate partly with each other because they can maximize the number of hydrogen bonds and reduce the repulsive interaction in this way. The examination of the results leads us to conclude that the hydration number of Na+ is 4, which agrees with the experiments[7]. The distance between Na+ and oxygen atom of water molecule in the first shell was computed by minimizing the TIPS2 potential function and the average value of the distance increased linearly from 2.286 Å to 2.324 Å as we went from the clusters with m=1 to m=7. This phenomenon was also found in other works[8] and in agreement with that obtained by SCF calculation[9].
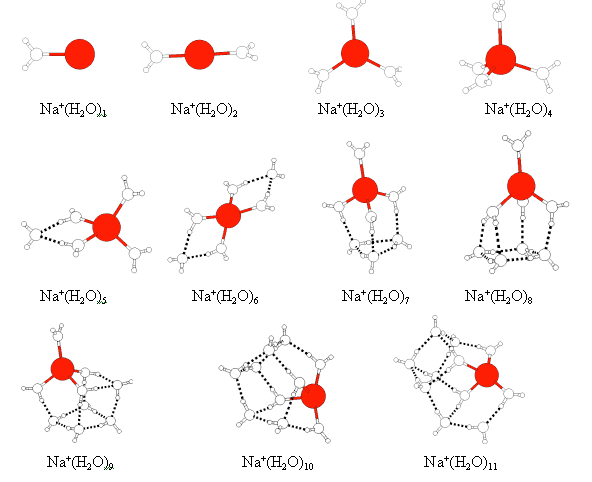
Figure 2 Geometry of the lowest energy structure found for Na+(H2O)m, m=1-11. Table 2 The potential energies of the Na+(H2O)m (kJ/mol )
| Na+(H2O)m | TIP | SPC | TIP3P | TIP4P | TIPS2 |
| Na+(H2O)1 | 98.5 | 101.9 | 103.6 | 93.0 | 94.7 |
| Na+(H2O)2 | 191.5 | 198.0 | 201.2 | 181.1 | 184.3 |
| Na+(H2O)3 | 274.0 | 282.6 | 286.9 | 260.0 | 264.2 |
| Na+(H2O)4 | 345.3 | 356.0 | 361.2 | 329.2 | 333.6 |
| Na+(H2O)5 | 396.0 | 415.1 | 416.9 | 386.7 | 390.4 |
| Na+(H2O)6 | 446.2 | 473.0 | 472.3 | 443.1 | 446.0 |
| Na+(H2O)7 | 483.7 | 517.2 | 515.7 | 489.0 | 491.2 |
| Na+(H2O)8 | 527.1 | 566.7 | 565.4 | 540.2 | 541.7 |
| Na+(H2O)9 | 567.4 | 617.5 | 612.0 | 595.4 | 595.6 |
| Na+(H2O)10 | 614.2 | 673.0 | 664.4 | 650.1 | 609.1 |
| Na+(H2O)11 | 658.7 | 724.7 | 714.6 | 696.1 | 695.0 |
Na+(OH2)n-1,n |
SPC |
TIP |
TIP3P |
TIP4P |
TIPS2 |
Lit [10] |
Lit [10] |
Expt [11] |
0,1 |
101.9 |
98.5 |
103.6 |
93.0 |
94.7 |
100.4 |
97.5 |
100.4 |
1,2 |
96.1 |
93.0 |
97.6 |
88.1 |
89.6 |
93.7 |
90.3 |
82.8 |
2,3 |
84.7 |
82.4 |
85.7 |
78.9 |
79.8 |
80.3 |
77.4 |
66.1 |
3,4 |
73.4 |
71.3 |
74.3 |
69.1 |
69.4 |
70.2 |
68.6 |
57.7 |
4,5 |
59.0 |
50.6 |
55.8 |
57.5 |
56.7 |
57.3 |
56.1 |
51.5 |
5,6 |
58.0 |
50.3 |
55.4 |
56.4 |
55.6 |
53.9 |
44.8 |
|
6,7 |
44.2 |
37.4 |
43.4 |
45.8 |
45.2 |
We have carried out potential energy minimization searches for the Na+(H2O)m (m=1-11) clusters using the hybrid algorithm and have succeeded in finding clusters global minimization potential energy structures using SPC, TIP, TIP3P, TIP4P and TIPS2 potential functions. The results show: using the five potential functions all reflect the potential energy properties of Na+(H2O)m clusters and gain almost the same geometries of Na+(H2O)m clusters. The hydration number of Na+ is 4, which is in agreement with the value of experiment. Acknowledgments The authors are grateful to Dr. J.A.Niesse (University of New Hampshire) for helpful suggestions. REFERENCES
[1] Niesse J A, Mayne H R. J. Comput. Chem., 1997, 18: 1233-1244.
[2] Gregurick S K, Alexander M H, Hartke B. J. Chem. Phys., 1996, 104 : 2684-2693.
[3] Moret M A , Pascutti P G, Mundim K C. J. Comput. Chem., 1998, 19 : 647-657.
[4] Aqvist J. J. Phys. Chem., 1990, 94 : 8021-8024.
[5] Jorgensen W L, Chandrasekhar J, Madura J D. J. Chem. Phys., 1983, 79 : 926.
[6] Jorgensen W L. J. Am. Chem. Soc., 1981, 103 : 335-340.
[7] Palinkas G, Radnat T, Hajdu F. Z. Naturforsch. Teil. A, 1980, 107: 35-44.
[8] Gowda B T, Benson S W. J. Phys. Chem., 1982, 86: 1544-1552.
[9] Clementi E, Popkie H. J. Chem. Phys., 1973, 58:1689-1699.
[10] Gowda B T, Benson S W. J. Chem. Phys., 1983, 79 : 1235-1241.
[11] Arshadi M, Yamdagni R, Kebarle P . J. Phys, Chem., 1970, 74: 1475-1483.