http://www.chemistrymag.org/cji/2004/06a070pe.htm |
Oct. 1,
2004 Vol.6 No.10 P.70 Copyright |
A density functional study of the adsorption of CO on Fe(111) surface
Chen Yunhong1, Yang Jun1,2,
Sun Yuchun1, Chen Manying1
(1Department of Chemistry, Jinan University, Guangzhou 510632 China; 2State
Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of
Sciences, Taiyuan, 030001)
Received on May 26, 2004; Support by the Committee of Science and Technology of China via 863 plan (No.2001AA523010)
Abstract The adsorption of CO on the Fe
(111) surface has been investigated by ab initio density functional methods and the
generalized gradient approximation (GGA). The surface is modeled using an infinite
periodic slab. CO adsorption at five different surface sites have been studied and it
shows that the energetically preferred site for CO adsorption corresponds to the shallow
hollow(a2)
adsorption followed by bridge site(a4), bridge-like site(a5), deep hollow site(a3), and on-top site(a1). However, CO is mostly activated in the bridge-like
site. The significantly increased theoretical CO bond length of 1.221Å on the
bridge-like site (compared to 1.144Å in the gas phase) indicates the activation of
CO bond and this site is expected to be the precursor for CO dissociation on the Fe (111)
surface.
Keywords Density functional methods; Fe(111) surface; carbon monoxide;
adsorption
1. INTRODUCTION
The adsorption of CO on transition metal low index surfaces has been the subject of
numerous experimental and theoretical studies in recent years because the CO adsorption
and dissociation play a crucial role in many catalytic processes, for instance, in car
exhaust or in the Fischer-Tropsch synthesis (FTS). As the iron-CO interaction plays an
important role in the FTS, the interaction of CO with clean iron surface has been
investigated by various techniques. There are many experimental and theoretical studies on
the Fe(001) and Fe(110) surfaces[1,2]. Both of these two surfaces are
close-packed surfaces, whereas, the Fe(111) surface is a very open surface. Difference in
the surface structure will affect the CO adsorption on the surface. There are relative few
studies on CO adsorption on the Fe(111) surface. Christmann and Seip [3] et al
have investigated the interaction of CO with an Fe (111) surface with low energy electron
diffraction (LEED). They found that the adsorption of CO on Fe (111) below 300 K present
three different non-dissociated species as distinguished by their C-O stretch frequencies.
According to this result, they thought that CO adsorbed on three different sites on the
Fe(111) surface, namely as "on top" ,
"shallow hollow" and "deep hollow"
sites. Whitemam et al [4] found that the CO
adsorption site occupation on the Fe (111) depends upon coverage and temperature, and CO
adsorbs on four sites on the Fe(111) surface. A new additional "bridge" site is found except the three sites mentioned above. Mehndru and
Alfred et al [5] have used semi-empirical molecular orbital theory to study the
binding and orientations of CO on Fe (100), Fe (110) and Fe(111) at low coverage. Their
investigation shows that CO binds on the di-s bridging site in the lying-down orientation on Fe (111) which do
not support the previous proposed "shallow-hollow" binding site for CO on Fe (111) surface[3]. It is
obviously that the level of the theory is insufficient and it can not be expected to give
a quantitative description of the process. Despite considerable valuable structure
information obtained from the experimental and theoretical studies described above, a
detailed mechanism and quantitative estimation of the energies of various processes of CO
interaction with the iron surface are still lacking.
In this paper, we present a systematic study of CO adsorption on the Fe
(111) surface at high coverage using ab initio density functional theory. The aims of the
present paper is to build up a detailed picture of the geometry, energetic and electronic
structure of the clean and adsorbed surface, and show some light in the CO adsorption
mechanism. The magnetism of the system and small energy differences between various spin
states make the study quite involved and challenging.
2 COMPUTATIONAL DETAILS
In the present study, all calculations described herein were performed within the
framework of density functional theory (DFT) using a basis set consisting of plan waves,
as implemented in the CASTEP package program [6] available in the Materials
studio 2.2 version of the Accelrys Inc. The electron-ion interactions were described by
ultrasoft pseudopotentials and electron exchange and correlation energies were calculated
with the Perdew, Burke and Ernzerhof formulation (PBE)[7] of the generalized
gradient approximation (GGA). Spin-polarization was included in the calculations for
systems with Fe to correctly account for its magnetic properties. Spin-polarization has
been shown to have a major effect on the adsorption energies for magnetic systems and may
alter the topology of the potential energy surfaces[8].
The surface of Fe (111) is modeled by a slab which contained seven
layers. The three topmost layers are allowed to relax, and CO is adsorbed only on one side
of the slab. The one-sided slab has been used intensively in the literature and has been
proven to be accurate. The plane-wave energy cutoff was set to 340 eV throughout all
calculations and the Brillouin zone of the surface unit cell was sampled with a 6× 6×1
Monkhorst-Pack mesh. The smearing width is 0.1eV. The coverage of the CO is defined as the
following: the coverage=the number of the CO molecule/the number of the surface Fe atoms.
CO can adsorb on the three topmost layers. So one CO adsorbed on the p(1×1) unit cell is
to represent the coverage of 1/3 ML. The adsorption energy per CO molecule is defined as:
Eads=E(slab+CO)-Eslab-ECO. In order to
distinguish the iron atoms in different layers, we labeled the iron atoms of the first
layer, second layer and third layer as Fe1, Fe2,Fe3,
respectively.
3.RESULTS AND DISCUSSIONS
To make sure that the GGA-PBE is suitable for the system of CO adsorption on the
Fe(111) surface, we calculated the CO bone length and a -Fe lattice constant and magnetic. The results listed in Table 1
show in agreement with the experiments results.
Table 1: Comparison between PBE and experiments
PBE |
Experiment |
||
CO |
Bond length (Å) |
1.14 |
1.13[9] |
a-Fe |
Lattice constant (Å) |
2.83 |
2.87[10] |
Magnetic moment (mB) |
2.24 |
2.22[10] |
This method has also been used to reproduce the results of CO adsorption on the Fe(001)[1] and Fe(110)[2], and the results show in agreement.
3.1 Clean surface
On the Fe (111) surface, an iron atom of the first layer is surrounded by three
shallow hollows and three deep hollows, shown in Fig. 1. A shallow hollow is formed by a
Fe2 atom and three Fe1 atoms (which are the nearest to the Fe2 atom), and the Fe2 atom
sits directly below the hollow. A deep hollow is formed by a Fe3 atom and three Fe1 atoms
(which are the nearest to the Fe3 atom), and the Fe3 atom sits directly below the hollow.
The very open structure of the clean Fe(111) surface is known to exhibit large multilayer
relaxation. With dik being spacing between the ith and kth atomic layer, and d
the bulk spacing. Sokolov et al [11] found the following relaxations: Dd12/d=(-16.9± 3.0)%, Dd23/d=(-9.8± 3.0)%, Dd34/d=(4.2± 3.6)%,and Dd45/d=(-2.2± 3.6)%. The
relaxations of our seven layer slab are shown in Table 2. Here only the three topmost
layers are relaxed and the relaxations are tabulated as relative changes in the nearest
neighbor distances di(i+3) instead of di(i+1).The agreement is
reasonable considering the uncertainties in the calculations due to the small number of
layers in the slab[12] and different k-points in the bulk and the Fe (111)
surface. The result shows that the first and second layers shifts inward and the third
layer shifts outward, and this agrees with the experiments qualitatively. The average
magnetic moment in the surface layer is slightly enhanced (ms=2.68uB)
compared to the bulk (ms=2.2uB), and this can be explained by the
open structure of the Fe(111) surface and the reduced coordination of the surface atoms.
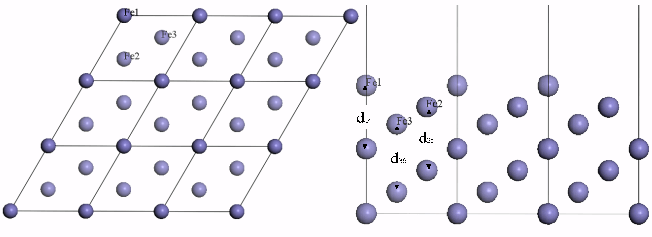
Fig.1 Top view and side view of Fe(111) surface
Table 2 Relaxation of the clean Fe(111) surface. The experimental data is from Ref [11]
DFT (%) |
Exp (%) |
|
Dd14/(3d) |
-2.5 |
-4.3 - -10.7 |
Dd25/(3d) |
-2.0 |
-2.6 |
Dd36/(3d) |
4.2 |
0.7 |
3.2 CO adsorption
As mentioned above, four possible adsorption sites on the Fe (111) surface at this
coverage have been experimentally detected. In our study, five adsorption sites are
identified, including a new bridge-like site, namely: on-top site (a1), shallow hollow site (a2), deep hollow site(a3), bridge site(a4), and bridge-like site (a5) (shown in Fig.2). On a1 and a2 sites, the CO molecules
are adsorbed perpendicularly to the surface. On the a3 and a4 sites, the CO molecules are found to be slightly
tilted to the surface. The two tilting angles are about 1oand 12owith respect to the
surface normal. The bridge-like site (a5) is a new adsorption side which has not been mentioned
both in experiments and theoretical studies. On this site, the carbon end of the CO is
bonded to two Fe1 atoms and one Fe2 atom. The oxygen atom is also bonded to a Fe1 atom and
the O-Fe bond length is 2.175Å. The CO tilting angle is 39° with respect to the
surface normal on this site. Table 3 summarizes the results for various sites. The results
indicate that at this coverage, CO prefers to adsorb on the a2 site, followed by the a4 site, a5 site, a3 site, and a1 site according to the adsorption energy of
these sites. The adsorption energy difference between the a2 site and the a4 site is very small, 0.02eV, and that between the a2 site and the a5 site is also very small
(0.1 eV). Both the adsorption energy and geometry of a2, a4 and a5 are very similar, these three adsorption sites may be
detected as one adsorption site by experiments. This is probably the reason that there are
three or four adsorption sites observed by experiments. The next question is to decide
which site to be the precursor for CO dissociation on the Fe (111) surface. Two factors
are considered: one is the adsorption energy which decides the appearance possibility of
the adsorption site; the other is the activation of the C-O bond (C-O bond length). Table
3 shows that the C-O bond length of a3 and a5 both are 1.221Å, but the adsorption energy of a5(2.33eV) is higher than
that of a3(1.79eV).
So the CO adsorbed on the bridge-like site is considered to be the precursor for CO
dissociation on the Fe (111) surface. On the a5 site, the oxygen atom bonding to a Fe1 atom also shows
the tendency to dissociate.
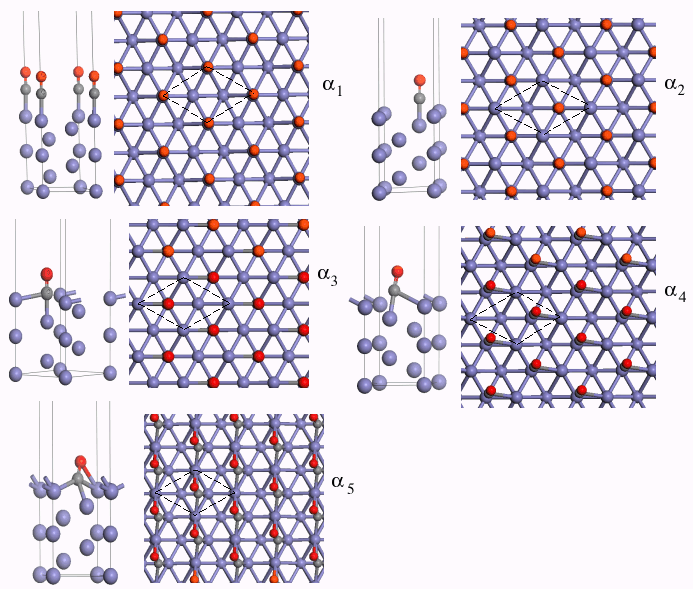
Fig.2 Side view and top view of a1, a2, a3, a4,
a5
Table 3 Results for CO adsorption on Fe(111) surface.
Site |
Eads(eV) |
dC-O(Å) |
dC-Fe(Å) |
a1 (atop) |
-1.54 |
1.160 |
1.828 |
a2 (shallow-hollow) |
-2.43 |
1.187 |
1.761 |
a3(deep-hollow) |
-1.79 |
1.221 |
1.853/2.334 |
a4 (bridge) |
-2.41 |
1.190 |
1.769/2.323 |
a5 (bridge-like) |
-2.33 |
1.221 |
1.824/2.111/2.128 |
Here Eads is referred to adsorption energy; dC-O, dC-Fe are referred to the bond length of C-O bond, and C-Fe bond
3.2.2 DOS analysis
A vast amount of literature has described the nature of the interaction between CO and
transition metals. It can be briefly summarized as follows: the occupied 5s orbital of CO
can readily overlap with the unoccupied dz2 and 4s orbitals of the
transition metal atoms with electron donation: the unoccupied 2p* orbitals of CO are also capable of overlapping with occupied dxz
and dyz orbitals of the metal atoms with electron back-donation[13].
The electronic density(DOS)is carried out in the text to analyze the binding mechanism. Fig. 3
shows the total density of states of the free CO and the partial density of states(PDOS)
of the C and O. The four peaks below E=0 are 3s 、4s 、1p 、5s (from the left to the
right) while the 2p*
antibonding orbital is about 7eV above the 5s orbital. These results agree with the
gas-phase photoelectron spectrum of CO qualitatively [14]. Comparison of the
DOS of the free CO molecule with the PDOS of C and O atoms shows that the components of
the 5s and 2p* orbitals are
mainly the C atom orbitals. That is the reason why in most cases the CO is adsorbed on the
metal surface with the C atom. Fig. 4 shows the PDOS of CO adsorbed on the five sites
where one can notice that both the 5s and 2p* orbitals are broadened over a range due to hybridization with the
iron orbital. The 2p*
orbital, empty in the gas phase, is now partially occupied upon adsorption, resulting from
interacting with the d-orbitals of the surface. The partial charge transfer leads to the
broadening of the 2p*
orbital with an edge below the Fermi level and the significant elongation of the C-O bond
due to the antibonding nature of the 2p* orbital. For all these adsorption sites, the 5s orbital intensity
is decreased dramatically. The PDOS of the CO adsorbed at various sites are very similar.
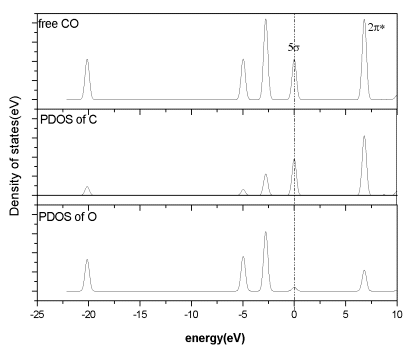
Fig.3 Total density of states of the free CO and the PDOS of the C
and O
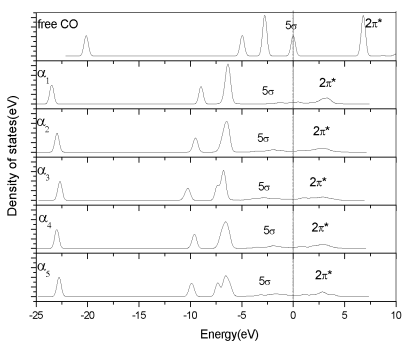
Fig.4 The PDOS of CO adsorbed on the five sites
4. SUMMARY
The interaction of CO with the Fe(111) surface have been studied by means of ab initio
density functional theory. Five adsorption sites are investigated including a new site
(bridge-like site) which is not discovered by the early experiments and theoretical
studies. The new site is expected to be the precursor for CO dissociation on the Fe (111)
surface. Our study finds that the CO molecule is mostly strongly bonded to the iron
surface at the shallow hollow, which is in good agreement with the early experiments. The
C-O bond is mostly activated in the bridge-like site. The binding mechanism is also
analyzed by using DOS analysis.
REFERENCES
[1] Nayak S K, Nooijen M, Bernasek S L J. Phys. Chem. B, 2001, 105: 164.
[2] Stibor A, Kresse G, Eichler A et al. Surf. Sci, 2002, 507-510: 99.
[3] Seip U, Tsai M C, Christmann K et al. Surf. Sci, 1984, 139: 29.
[4] Whitman L J, Bartosch C E. J. Chem. Phys, 1989, 90: 2050.
[5] Mehandru S P, Anderson A B. Surf. Sci, 1988, 201: 345.
[6] Payne M C, Teter M P, Allan D C et al. Rev. Mod. Phys, 1992, 64 (4): 1045.
[7] Perdew J P, Burke K, Ernzerhof M. Phys. Rev. Lett, 1996, 77: 3865.
[8] Ge Q, Jenkins S S, King D A. Chem. Phys. Lett, 2002, 327: 125.
[9] Herzberg G. Molecular spectra and Molecular structure. I. Spectra of Diatomic
Molecules, 1950, p.521
[10] Kittel C. Introduction to Solid State Physics, New York: Wiley, 1996.
[11] Sokolov J, Jona F, Marcus P M. Phys. Rev B, 1986, 33: 1397.
[12] Mortensen J J, Ganduglia-Pirovano M V, Hansen L B. Surf. Sci, 1999, 422: 8.
[13] Hansong C, David B R, Sheldon W D. J. Phys.Chem, B, 2001, 105: 12547.
[14] Plummer E W, Salaneck W R, Miller J S. Phys. Rev. B, 1978, 18: 1673.