http://www.chemistrymag.org/cji/2004/06b076pc.htm |
Nov. 1,
2004 Vol.6 No.11 P.76 Copyright |
Study of the catalytic performance and stability of complexes with uncoordinated atoms upon methanol carbonylation
Ling Chen, Shao Shou-yan, Qian Qing-li#,
Pan Pin-glai#, Yuan Guo-qing#*
(Jiangsu Suopo Group Co. Ltd., Zhenjiang 212006; Institute of Chemistry, Chinese Academy
of Sciences, Beijing 100080)
Abstract 2-methyl butanoic acid
pyridine and b-(2-pyridyl)
ethyl succinate were used as ligands to form square planar rhodium(I) cationic complexes
of cis-dicarbonyl coordinated bidentate with tetracarbonyldichlorodirhodium. Due to the
uncoordinated N and O atoms in the above ligands, which can substitute the terminal
carbonyls to form new coordinating bonds when heated in the absence of carbon monoxide,
the stability of the complexes increase. The results show that such complexes with
uncoordinated atoms have the same catalytic performance with simulating complexes without
uncoordinated atoms. Moreover, they demonstrate peculiar reaction stability, which is the
characteristic of intramolecular bond substation reaction.
Keywords uncoordinated atom; catalysts; intramolecular substitution reaction;
methanol carbonylation
(江苏索普(集团)有限公司,镇江 212006; 中国科学院化学研究所,北京 100080) 摘要 本文分别以4-(2-吡啶基)丁酸甲酯及丁二酸二[2-(2-吡啶基)]乙酯为配体,与四羰基二氯二铑形成正方平面鳌合型顺二羰基铑阳离子配合物。丁二酸二[2-(2-吡啶基)]乙酯铑配合物中未参与配位的N、O原子在无一氧化碳存在下遇热可取代铑的末端羰基形成新的配位键,提高了其稳定性。研究结果证明,该类含有未配位原子的配合物在催化甲醇羰基化制备乙酸的反应中,不仅具有与不含未配位原子的模拟配合物相同的催化性能,而且由于存在分子内键取代反应,表现出独特的反应稳定性。
关键词 未配位原子;催化剂;分子内取代反应;甲醇羰基化
甲醇羰基化反应制备乙酸,其经典的催化剂活性物种为铑的
[Rh(CO)2I2]-结构形式[1]。[Rh(CO)2I2]-物种虽然具有良好的催化活性,但在反应物分离部分由于一氧化碳的分压降低,一价铑易变为三价铑[Rh(CO)2I2]-而发生沉淀,限制了反应体系中铑浓度的增加,进而影响其相应生产效率的提高。为了克服上述铑催化剂的不足,多年来大量的研究工作围绕以下内容进行:1. 选用含有N、O、P、S授体原子的小分子或高分子为配体与铑形成新型结构的配合物[2-7],来提高反应的催化活性及稳定性。2. 在反应体系中添加各种金属盐和其它类型的化合物以提高催化体系的反应速率[8] 。在为数众多的催化剂研究中,含有未配位原子的正方平面顺二羰基铑配合物显示出明显的优势。研究发现,这类正方平面顺二羰基铑鳌合型阳离子配合物在遇热条件下,空余的未配位授体原子可取代铑的末端羰基而与铑形成新型配位键。这种新型的配位形式在一氧化碳存在时即可回到最初的配位状态。这一特征使得该类配合物在空气中的热稳定性得到极大的提高[9]。
本文在前期工作基础上,运用实验手段,进一步探讨和证实了该类化合物催化甲醇羰基合成反应的稳定性,并设计了与其结构相似的模拟配合物进行对比。结果证明:该类配合物的分子内键取代反应在无一氧化碳保护的情况下仍可发生作用,这种键取代反应的独特性能使得Rh处于活性价态,从而大大提高了其作为催化剂的稳定性。
1 实验部分
1.1 配合物的制备
称取1摩尔份的丁二酸二[2-(2-吡啶基)]乙酯,溶解于甲醇中于0℃下搅拌滴加含有0.5摩尔份四羰基二氯二铑的甲醇溶液。反应10分钟后加入四苯硼钠水溶液,待出现沉淀后,过滤,室温真空干燥至恒重,得到柠檬黄色粉末状配合物。
4-(2-吡啶基)丁酸甲酯铑配合物按同样方法制备。
1.2 配合物的表征
配合物的IR数据由Bruliet
EQUINO×55红外光谱仪测定;XPS数据由ESCALAB220i-XLX射线光电子能谱仪测定,激发源:AL
Ka15KV×200mA。
1.3 配合物的性能评价
催化反应在100ml反应釜中进行,反应产物用GC-8810型气相色谱仪鉴定。
2 结果与讨论
配合物的结构如下:

配合物(Ⅰ) 配合物(Ⅱ)
由于配合物
(Ⅱ)含有未配位的N、O两个授体原子,其在遇热条件下可发生下列反应[10]:

配合物
(Ⅱ) A 配合物(Ⅱ)B 配合物(Ⅱ)C将上述键取代反应的表征数据列于表
1中。如表1所示,配合物铑的末端羰基特征吸收峰在1900-2100 cm-1范围,其中配合物(Ⅰ) 的特征峰在1990,2080cm-1,配合物(Ⅱ)在2000,2100cm-1 。 将配合物在空气中于100℃加热30分钟后, (Ⅰ) 的红外特征峰消失,证明配合物已分解,而配合物(Ⅱ)却在1950 cm-1处出现一个新的铑末端羰基吸收峰,为其处于(Ⅱ)B结构时的振动吸收,即铑的顺二羰基中有一个羰基被配体中的空配位授体原子所取代。随着加热时间的延长,新的羰基吸收峰逐渐加强,初始的两个吸收峰不断减弱,说明结构(Ⅱ) A为结构(Ⅱ)B所取代。继续加热升温,位于2000,2100cm-1处 的初始峰及1950 cm-1处的羰基峰全部消失,说明铑的两个末端羰基被全部取代,(Ⅱ)B转化为(Ⅱ)C结构。将结构(Ⅱ)C在50℃加热的条件下通入CO时,配合物(Ⅱ)的初始吸收峰重新出现,又恢复到初始状态 (Ⅱ) A,证实了含有未配位授体原子铑配合物分子内取代反应的可逆性。 表1 配体和配合物的IR和XPS数据Table 1 IR and XPS data of ligand and complexes
Ligand and |
Binding Energy Eb /eV |
|||
N1S O1S Rh3d5 |
||||
[Rh(CO)2Cl]2 ligand (I) |
2082 2018 |
532.1 |
311.2 |
|
| complex (I) | 1990 2080 | 398.3 | 532.1 | |
| complex (II)A | 2000 2100 | 399.8* | 533.0 | 308.7 |
| complex (II)B | 1950 | 399.8* 398.7 | 533.4 532.7 532.2 | 309.8 |
| complex (II) C | 2000 2100 | 399.8* 398.8 | 533.4 532.6 532.0 | 309.4 |
| after disposal of CO | 399.8* | 533.6 532.5 532.0 | 308.8 | |
表
1的XPS能谱变化同样证实了上述配合物的结构和分子内键取代反应的发生。配合物(Ⅰ) N1S的结合能在XPS谱图上只表现出一种399.8 eV配位状态,比其配体的N1S升高了1.5eV。配合物(Ⅱ) A的N1S原子则表现出配位及未配位两种状态,其中已配位N1S 为399.8 eV ,比其未配位N1S的398.7 eV升高了1.1eV;当其转变为(Ⅱ)B结构时,由于只是与中心铑原子相邻的O原子取代了一个羰基,氮原子的状态并未发生改变,所以氮原子的能谱峰也未发生变化;但当配合物转变为(Ⅱ)C结构后,能谱图上只剩下结合能为399.8eV的N原子,证明配合物(Ⅱ)C中未配位的N消失、N原子已全部成键。在氧的光电子峰中,532.0~532.2 eV的光电子峰为配合物中未配位氧与铑的末端羰基氧,在533.4 eV以上处为-O-,而532.5~532.7 eV处则为与铑配位的O1S光电子峰,其中532.0~532.2 eV 的光电子峰包括样品中的污染氧[10,11]。由表1可以看到,配位氧与未配位氧的电子结合能子相比,升高了0.5~0.6 eV。随着配合物(Ⅱ)A® B® C的变化,其参与与铑配位氧的O1S光电子峰强度不断增加,表明配体中的羰基氧逐步完成与铑原子的配位成键。
配合物(Ⅰ)、(Ⅱ)中的铑原子,由于接受了从N和O转移来的电子,其Rh3d5结合能与[Rh(CO)2Cl]2中的311.2eV相比均有所降低。 配合物(Ⅰ)下降到308.7eV;配合物(Ⅱ)A 比[Rh(CO)2Cl]2下降了1.4 eV为309.8 eV,当其转化为结构(Ⅱ)B时结合能降为309.4 eV,进一步转化成结构(Ⅱ)C后,其结合能下降为308.8eV。上述配合物的电子结合能的变化,反映了其原子状态在加热或通CO处理时发生了改变,同时与红外光谱分析相吻合。
在这类铑配合物中N、O原子是电子施体,铑原子是电子的受体。羰基与金属配位形成dπ-pπ反馈键,结果在d电子电荷分布中造成空隙,使配体更容易接近。据此,当二齿N、O配体接近铑原子时,N、O原子极易将其孤对电子填入铑的空d轨道,与铑原子结合为s 键,而反馈成键的可能性却很小[12,13]。显然,空配位的授体原子N、O与Rh所形成的N® Rh、O® Rh配位键,可以取代Rh与其末端羰基形成的较强的Rh® Cp 反馈键;在一氧化碳气氛下,Rh® Cp 反馈键可重新取代N® Rh、O® Rh键,使配合物又回复到顺二羰基铑的初始结构;但不含空配位授体原子的结构相似的铑配合物,其稳定性相对较差。
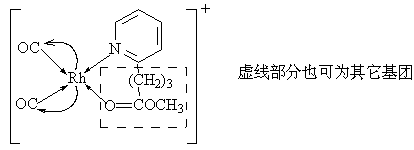
羰基铑配合物是一种用途广泛的催化剂,但遇热易分解是这类配合物的共同弱点,而造成其不稳定分解的主要因素是铑的末端羰基易脱离致使其处于配位不饱和状态。选用含有空配位授体原子的配合物,在遇热时其未配位原子可迅速与铑配位取代脱落的羰基,因此其在空气中的热稳定性通常可以提高到100℃以上[14],同时还避免了反应过程中由于高温引起的螯合结构的破坏而使铑变价失活。这一分子内弱键取代较强配位键的反应在理论上有着重要的意义,其实际应用价值有待于逐步验证。
2.2 甲醇羰基化反应
正方平面顺二羰基铑配合物是催化甲醇羰基化制备乙酸反应的典型催化剂活性物种。图1是(Ⅰ)、(Ⅱ)两种配合物催化甲醇羰基化反应体系中各组分产率及转化率随反应时间的变化关系。

图
Fig 1 Relationship between each composition and reaction time.
(reaction conditions: CH3OH 0.247mol; CH3I 0.04mol; Rh 7.16×10-5mol; PCO=3.0MPa; T=140℃)
由图
1可知,配合物在催化甲醇羰基化反应制乙酸时,通常短时间内单釜的催化反应活性基本上没有差异,即配体不同对碘甲烷在羰基化反应中的加成步骤没有影响,该反应中间活性物种CH3COI的生成也不因配体的不同而变化。甲醇羰基合成乙酸的羰基铑催化剂,在反应中由于一氧化碳保护不足或遇到空气时,一价铑易变为三价铑而处于非活性价态。通常在碘化氢存在的乙酸介质中,并且在一氧化碳压力为1.0Mpa时,三价铑可于反应温度下回复到一价的活性态。即使如此,铑的易变价失活及形成三价铑沉淀仍是工业生产中的一个难点,因为在产物与催化剂分离的闪蒸阶段,一氧化碳分压与反应压力相比大大降低,这样当一氧化碳保护不足时,便影响了催化剂的循环使用效率。图2的实验,将这两种配合物在反应过程中对空气的稳定性进行了比较。

图2 不同配合物在反应体系接触空气前后催化活性的比较
Fig 2 Comparison of catalytic activity of different complexes before and after the reaction system was exposed to air.
(reaction conditions:CH3OH 2.97mol; CH3I 0.48mol; Rh 6×10-4mol; PCO =4.0MPa; T=140℃)
如图
2所示,(Ⅰ)、(Ⅱ)两种配合物在催化甲醇羰基化反应进行到20分钟时停止反应并冷却,将反应液倾入烧杯中并使其暴露于空气中30分钟,然后继续装入压力釜反应。实验结果表明,配合物(Ⅱ)与配合物(Ⅰ)相比具有明显的优势,因其发生了分子内取代反应而将铑保护在活性价态,再次进入反应体系时可很快进行催化反应;配合物(Ⅰ)因暴露于空气中致使铑变为高价态失活,尚需要有一个重新活化的过程才能再次转为活性状态。图3是(Ⅰ)、(Ⅱ)两种配合物在10次对照试验中其甲醇转化率的比较。实验程序为:反应至1小时后,将反应液放入蒸馏分离塔中,在氮气保护下将2/3反应液蒸出,然后用一氧化碳将蒸馏后所剩1/3含催化剂的母液及新加入的甲醇、碘甲烷混合液压回到反应釜中,继续进行反应。

图3甲醇转化率与反应时间的关系 (10次对照试验,每次一小时)
Fig 3 Relationship between methanol conversion rate and reaction time (comparing reaction for ten times, one hour for each time)
(reaction conditions:Rh 4.5×10-4mol; CH3OH/CH3I=10 (Mole ratio); T=135℃; PCO=3.2MPa)
由图
3可以看出,配合物经10次反应对照,每次反应后配合物(Ⅰ)的活性与配合物(Ⅱ)相比略有下降,反应10次后已经下降了5%左右。这说明配合物(Ⅰ)在无一氧化碳保护的条件下分离时,有部分铑配合物的末端羰基脱落而导致催化剂分解。显然,配合物(Ⅱ)在很大程度上避免了这种情况。 REFERENCES[1] Haynes A, Mann B E, Gulliver D J, et al. J. Am. Chem. Soc., 1991, 113 (22): 8567-8569.
[2] Chen Yuying, Yuan Guoqing, Wang Dianxun et al. J. Mol. Cat. (Fenzhi Cuihua), 1988, 2 (3): 147-153.
[3] Jonathan R. Dilworth, John R.Miller, Nigel Wheatley, et al. J.Chem.Soc. Chem.Commun., 1995, 1579-1580.
[4] C.A.Carraz, E.J.Ditzal, A .G.Orpen, et al. Chem.Commun., 2000, 14: 1277-1278.
[5] Das P, Boruah M, Kumari N., et al. J. Mol. Catal., 2002, 178: 283-287.
[6] Nandini Kumari, Manab Sharma, Pankaj Das, et al. Appl. Organometal. Chem., 2002, 16: 258-264.
[7] Thomas C M, Süss-Fink.. Coordin. Chem. Rev., 2003, 243: 125-142.
[8] Murphy M A, Smith B L, Torrence G P, et al. J. Organomet. Chem., 1986, 303 (2): 257-272.
[9] Peng Wen-qing,Yuan Guoqing. CHINESE SCI BULL (Kexue Tongbao), 1996, 42 (9): 935-937.
[10] Peng Wen-qing,Yuan Guo-qing. CHINESE SCI BULL (Kexue Tongbao), 1997,42 (16): 1729-1731.
[11] Wang Dang-han(,Wang Min-gui,Liu Shi-hong. Vacuum Science and Technology (Zhikong Kexue Yu Jishu)., 1984, 4: 1-4.
[12] Fordyce W.A., Crosby G.A. Inorg. Chem., 1982,21:1023-1026.
[13] Aresta M., Defazio M. J. Organomet. Chem., 1980,186:109-120.
[14] Jiang H, Diao K S, Pan P L, et al. Chin. J. Chem., 2000, 18 (5): 751-755.