http://www.chemistrymag.org/cji/2005/071002rc.htm |
Jan.1, 2005 Vol.7 No.1 P.21 Copyright |
Jiang Dazhi
(Institute of Chemistry, Chinese Academy of Sciences, Beijing 100080) Abstract Progress on the synthesis of acetic acid and anhydride by the catalyzed carbonylation, particularly during the last fifteen years or so was summarized.
Keywords Acetic acid Acetic anhydride Carbonylation Synthesis Catalysis 乙酸和乙酐羰基合成及其催化技术 蒋大智
(中国科学院化学研究所 北京100080) 摘要 综述了羰化法合成乙酸和乙酐及其催化技术的研究进展、特别是近10多年来的新发展。
关键词 乙酸 乙酐 羰化 合成 催化
作为一类重要的基础有机化工原料,乙酸和乙酐及其下游衍生产品,被广泛地应用于医药、合成纤维、轻工、纺织、皮革、农药、炸药、橡胶和金属加工、食品以及精细有机化学品的合成。
工业上合成乙酸和乙酐的起始原料最初是粮食,后来转向矿石、木材、石油、煤炭和天然气。国外乙酸生产当前主要采用乙醛氧化法、烷烃和轻质油氧化法、以及羰化法(羰基合成法)。其中低压甲醇羰化法是上世纪60年代末开发出来的技术上最先进的方法。采用此法生产的乙酸现已占乙酸总生产量的70%以上。而乙酐的工业生产,目前国际上普遍采用的是乙醛氧化法、烯酮法和羰化法,其中羰化法制乙酐是上世纪70-80年代在低压羰基合成法制乙酸基础上发展起来的新工艺,用该技术生产的乙酐现已占到世界总产量的45%以上。本文简要评述乙酸和乙酐羰基合成工艺路线及其催化技术。
采用羰化法合成乙酸和乙酐,起始原料是一氧化碳、甲醇、二甲醚或其它甲氧基化合物。反应热力学计算和实验测定表明,羰化反应是放热反应。但由于反应活化能高,必须在催化剂的催化作用下才有可能实现这一反应过程。正因为如此,羰基合成法生产乙酸和乙酐的核心课题一直是高性能的催化剂体系及其相应的工艺技术的研究[1]。
羰基合成法生产乙酸和乙酐的催化剂体系及其相应工艺,总体来说,可以分为两大类,即酸性催化剂体系的Koch碳正离子羰化反应路线和金属羰基化合物/卤素催化剂体系的Reppe络合催化羰化反应路线。本文对这两种羰化反应路线的发展历史分别予以概述,并重点评述以络合催化羰化反应路线进行的乙酸和乙酐羰基合成及其催化技术近10多年来的新发展。
1 Koch羰化反应工艺
20世纪20年代, Celanese公司的H.Dreyfus以BF3/H3PO4为催化剂,用铜和银作助催化剂,发现甲醇和一氧化碳(CO)在310℃高温和200
MPa高压条件下可直接反应生产乙酸,并于1925-1928年进行了100 kg/d的中间试验[2]。由于反应液腐蚀性极强,当时使用的反应釜以黄金做衬里,测得甲醇转化率为40%,选择性为70%。后来因为1929年的世界经济大萧条,
试验未能继续下去。到了20世纪50年代Koch反应概念确立后,进一步的实验表明[3],在BF3/H2O催化体系下,由甲醇到乙酸的羰基合成的反应温度可控制在433-573K之间,CO压力可降低到10
MPa左右,乙酸收率可达到92 %。
研究表明,在采用磷酸、硫酸或氢氟酸等无机酸和三氟化硼、三氯化铝等Lewis酸/水催化体系条件下,甲醇羰化合成乙酸的反应,首先有甲基碳正离子H3C+
生成,继而与CO反应形成乙酰基碳正离子H3CC(O)+,再与水反应,形成乙酸。质子H+
或电子对的受体物质是反应的催化活性物种。由此可见,在这一反应过程中,含水的酸性催化剂体系是必不可少的。通常所使用的酸性催化剂,除上述无机酸和Lewis酸外,也有使用固体酸等超强酸类物质的文献报道[4-5]。Cu(I)
和Ag(I) 等IB族金属离子,经常作为助催化剂使用。因为这类金属离子很容易与CO形成多羰基配合物,有助于将CO从气相转移到反应界面或反应介质内,并向反应提供活化态的CO。鉴于碳正离子中间体的形成在这一反应过程中起着重要作用,这一反应更有利于被用来制备分子骨架中羧基接点碳上支化度高的有机羧酸,特别是支链C6~C11羧酸。例如,采用Cu(I)
或Ag(I)/浓硫酸催化剂体系,可以在常温常压条件下生产叔碳酸[6]。而对于由伯醇合成伯羧酸,其难度或者说反应条件则相对地要苛刻一些。
采取Koch碳正离子羰化反应进行乙酸的羰基合成,其羰化过程通常需要维持在较高的反应温度和较高的一氧化碳分压环境下,与下面将要讨论的Reppe络合催化羰化反应工艺相比,在反应条件和设备材质要求方面显现不出什么优越性。由于在Koch羰化反应过程中要有碳正离子的生成,其反应温度一般地说还要更高一些;特别是其羰化反应速率和产物选择性远不如后者。但它的一个重要优点是,不需要使用碘甲烷和碘盐[7],甚至可以不使用卤素或卤代物[8]。这是迄今在乙酸和乙酐羰基合成领域,人们还非常重视将Koch羰化反应路线作为重要课题研究的原因。
1984年K.Fujimoto等[4] 使用固体酸催化剂在573-673K和1.0
MPa气-固相催化反应条件下,使甲醇羰化合成乙酸和乙酸甲酯。采用气-固相催化羰化反应,反应压力显著降低,但羰化产物收率极差。A.Calafat等[9]
使用硫化的钴-钼/活性碳(Co-Mo/A.C.)催化剂,在无任何促进剂条件下也是采用气-固相催化羰化反应,由甲醇合成出乙酸和乙酸甲酯,甲醇转化率20%,羰化选择性50%左右,时空产率
0.15 mol/kg-cat.h。彭峰对此作出改进[10],使用的催化剂不再含VIII族金属钴,而是硫化钼/活性碳(MoS2.5/A.C.)催化剂床,反应压力100
kPa,温度573K,原料气空速3000 L/kg-cat.h,甲醇摩尔浓度23%,在优化反应条件下,甲醇转化率50%,羰化产物是AcOMe,选择性80%,时空产率为
6.16 mol AcOMe/kg-cat.h。钼在催化剂中占14 wt.%,故相应的催化反应速率为4.2
mol AcOH/mol Mo.h。尽管这一结果还远不能与Reppe反应结果相比[11],但毕竟拓展了乙酸羰基合成的一个新的发展方向。
应用Koch碳正离子羰化反应合成乙酐,初始原料无论是甲醇或二甲醚,都要先转化成乙酸甲酯。在酸性催化剂体系催化条件下,由乙酸甲酯到乙酐的羰化反应,同样有甲基碳正离子和乙酰基碳正离子作为反应中间体形成,乙酰基碳正离子随后进一步反应,形成乙酐。反应的催化活性物种同样是质子H+
或电子对的受体物质。有关利用Koch羰化反应进行乙酐合成的报道,文献中所见甚少,原因可能在于这样的酸性催化剂体系很难找到。由于乙酐的羰基合成需要无水环境,而所有的强酸性物质、特别是无机酸,在无水条件下,是很难提供催化活性物种的。美国Texaco公司在20世纪80年代曾采用不含碘的催化剂,由乙酸甲酯合成乙酐[12]。他们所使用的催化剂为水合RuO2/季鏻盐/Co2(CO)8,其中季鏻盐为四正丁基鏻溴化物(n-Bu4PBr)。在结果最好的实验中,反应温度维持在220℃,总压为39
MPa,经过18小时反应,乙酸甲酯转化率为72%,乙酐选择性为64%,反应体系中的水含量测定值为0.02%。发现在上述催化剂中,如果不加羰基鈷,则催化活性极低。由此看来,使用水合RuO2/n-Bu4PBr/Co2(CO)8催化剂,由乙酸甲酯和一氧化碳合成乙酐,其中起主要作用的究竟是Koch羰化反应、还是Reppe羰化反应,还很难说。
2 Reppe羰化反应工艺
1941年德国BASF公司的Reppe等[3,13]发现,VIII族金属羰基化合物在有卤素或卤化物存在的情况下,甲醇羰化反应可在250-270℃和20-50
MPa压力下进行,当时所试验的三种VIII族金属的催化活性顺序是:Ni>Co>Fe。BASF公司在此基础上成功地开发出高压法钴/碘催化剂的乙酸羰基合成工艺,反应温度250℃,压力53MPa,产物以甲醇计收率为90%,以CO计为70%。1960年在德国Ludwigshafen建成3600
t/a的生产装置,反应釜材质用哈氏合金B,生产能力后来扩大到45 kt/a。并以此规模向罗马尼亚和美国各转让了一家,其中美国Borden
Chemical公司于1966年采用此工艺在路易斯安娜州建成的一套装置,其生产能力1981年扩大到64
kt/a。由于操作压力过高等条件限制,生产规模很难做大。1980年代BASF公司专利报道采用Co/MeI催化剂的乙酸羰基合成[14],反应温度180℃,压力70
MPa,催化反应速率达到11.6 mol AcOH/mol Co.h。Reppe等[15,16]还用镍、钴等羰基化合物/卤素催化体系由乙酸甲酯羰化合成乙酐,反应温度190℃,压力60-70MPa,得到镍、钴催化剂的催化反应速率分别为2.9
mol AcOAc/mol Ni.h和5.8 mol AcOAc/mol Co.h。这些早期的结果此后很快便被刷新,不仅催化活性显著提高,而且反应条件、特别是反应压力被有效地降低。用于乙酸和乙酐羰基合成的其它VIII族金属或双金属催化剂,以及VIII族金属与其它族过渡金属元素组合的双金属或多金属催化剂,亦被广泛地进行了实验和研究。
1968年美国Monsanto公司的F.E.Paulik和J.F.Roth [17]发现,在用于乙酸羰基合成的VIII族金属元素中,铑催化剂有着更高的催化活性和选择性,反应条件也十分温和。采用铑化合物/碘甲烷催化剂的均相溶液法工艺,由甲醇合成乙酸,反应温度可降到175-200℃,压力降到6.8
MPa以下,催化反应速率达1.1×103 mol AcOH/mol Rh.h,羰化选择性大于99%,从而被冠以“低压法”之美名,为近代乙酸羰基合成工业树立了一块里程碑。1970年由该公司在Texas州建成135
kt/a的生产装置[18]。1975年生产能力提高到180 kt/a,其后又提高到272
kt/a。低压羰化法合成乙酸具有明显的技术和经济优势,自1970年工业化以来,在全球各地建立起来的低压羰基合成大型装置的生产能力占乙酸工业新增生产能力的90%以上。
Reppe羰化反应是一类络合催化反应过程。研究表明,许多过渡金属化合物都能用作为这类羰化反应的催化剂,其中真正的催化活性物种是这些过渡金属的羰基化合物[3]。作为羰化反应催化剂的主体金属,目前被研究最多的是周期表中Ⅷ族过渡金属。就单一主体金属的催化活性、选择性、以及温和的反应条件而论,铑金属化合物独占鳌头。由于在Reppe羰化反应体系中,作为催化活性物种的Ⅷ族金属羰基化合物的组成和结构各异,催化羰化反应机理和反应动力学各不相同。限于篇幅,本文仅对文献中报道较多的均相溶液铑/碘催化剂体系的催化羰化反应进行讨论。Paulik和Roth等最初的实验结果[18,19]及后来的研究都表明,在甲醇羰化反应中,采用包括金属铑粉在内的各种0-3价铑的物种作为催化剂主体金属的前体,经过不同的反应处理和一定的反应诱导期,它们都能对羰化反应表现出类似的高催化活性和高选择性。Forster等研究认为[20,21],在CO和碘负离子存在的条件下,这些化合物都可转化为具有催化活性的[Rh(CO)2I2]-,后者具有正方平面构型。由于铑原子与CO配位基之间以σ-π成键及反位效应,其顺式结构较为稳定。在以卤代甲烷为助催化剂的羰化反应中,羰化反应实际上是通过助催化剂卤代甲烷、特别是碘甲烷对催化活性物种[Rh(CO)2I2]-的氧化加成等反应生成乙酰卤化物而实现的:
CH3X + CO ![]() CH3-CO-X X=Cl, Br, I
CH3-CO-X X=Cl, Br, I
当反应体系中存在有大量甲醇和水时,生成的乙酰卤化物与之反应,便生成乙酸或乙酸甲酯,同时再生成卤代甲烷或卤化氢。卤化氢与甲醇反应再被转化成卤代甲烷,继续被重新使用。作为助催化剂的卤素或卤化物,其助催化作用有如下顺序,即I>Br>Cl。在主要以碘甲烷为助催化剂的均相溶液反应体系中,且在充分供给反应原料甲醇和一氧化碳的情况下,甲醇羰化反应动力学研究指出[18,20-22],碘甲烷对[Rh(CO)2I2]-的氧化加成是羰化反应过程的速度控制步骤。催化反应速度
r = k.[Rh].[MeI],式中[Rh]和[MeI]分别为反应体系中铑物种和碘甲烷的浓度。羰化反应为二级反应。Hjortkjaer和Jensen[22]由实验测定甲醇羰化反应在150-225℃的温度范围内的活化能Ea和速度常数的指前因子k0分别为61.5
kJ.mol-1和3.5×106 L.mol-1.sec-1,反应活化热焓△H≠=56.9 kJ. mol-1,活化熵△S≠= -134.8 J.mol-1.K-1。
对络合催化羰化反应活性物种的研究表明,铑金属离子必须处于最低氧化价态,即一价铑Rh(I)才具有催化活性。一价铑配位活性物种在失去CO等还原性气体或惰性气体保护时,很容易被氧化分解为RhI3而沉淀析出。为保持催化活性物种的稳定,有文献报道[23]采用含有两个或以上具有孤对电子的有机分子螯合铑金属离子、或者采用碱金属化合物,以防止与铑活性物种负离子配对的正离子的缺位。还有一种简便办法是,往反应体系中加入氧化电位较高的处于低价态甚至零价态的可变价金属或其化合物添加剂,例如低价态甚至零价态的锡盐、铜盐或铬盐等,或者它们的羰基化合物。实验表明[24],添加适量SnCl2对催化剂的活性没多大影响,但却能明显地阻止铑催化剂在CO分压降低时分解现象的出现。
20世纪70-80年代,由于石油危机和Monsanto合成乙酸工艺成功范例的启示,国际上纷纷开展了低压均相溶液法羰化合成乙酐的实验和研究,其中日本Ajinomoto[25]和Showa
Denko[26]、美国Halcon[27,28]和德国Hoechst[29-31]几家公司均有专利报导。1973-74年,这几家公司采用铑基催化剂体系对乙酸甲酯进行羰化合成乙酐的实验,结果表明羰化反应压力可降低到3-10
MPa,反应温度为150-250℃。1983年Halcon公司与 Eastman-Kodak公司所属的Tennesse公司合作,在Kingsport建成世界上最大的乙酐厂,年生产能力为22.7万吨,一次投产成功[32]。反应温度为150-200℃,压力为1-10
MPa,羰化选择性在99%以上,连续实验的催化反应速率为500 mol AcOAc/mol
Rh.h[33-35]。该厂运转几年后,证明经济上有吸引力[36]。英国BP公司1982年引进Monsanto技术建成了一套170
kt/a的羰化法制乙酸生产装置后,一方面致力于将设备能力提高到235
kt/a,另一方面又在Monsanto法制乙酸和Halcon法由乙酸甲酯制乙酐技术的基础上,开发出由甲醇羰化联产乙酐和乙酸的新流程[37],于1987年在Hull建厂,1989年建成投入运行,年产乙酐6.5-10.5万吨,乙酸7.5-12万吨。1991年Halcon公司又建成规模更大的羰化法乙酐生产装置,并且参照英国BP公司的做法,改起始原料乙酸甲酯为甲醇,年生产乙酐27万吨,并联产乙酸8万吨。
有文献[38]用Ⅷ族的Ir、Os、 Pt、Pd、Rh、Ru等过渡金属化合物作催化剂,在含有氮、磷原子基团的有机化合物及碘甲烷存在下,对乙酸甲酯羰化合成乙酐进行了研究,反应温度195℃,CO分压3
MPa,结果表明,Rh催化剂活性最高,是Ir的5倍,Ru和Pd是低活性过渡金属,PtCl2、OsO4、NiCl2的活性最低,催化活性顺序是:Rh>Ir>Ru>Pt>Os>Ni。在络合催化羰化合成乙酐反应过程中,碘甲烷对
[Rh(CO)2I2]-的氧化加成生成乙酰卤化物的催化循环与乙酸羰基合成是一样的[1],只是乙酐羰基合成要求反应体系基本上应是无水或少水体系。在乙酐羰基合成工艺中,由于体系内存在有大量的乙酸甲酯和乙酸盐,产生的乙酰卤化物与之反应形成乙酐,同时再生成卤代甲烷或卤盐。卤盐与乙酸甲酯反应再被转化成卤代甲烷和乙酸盐,而被继续重新使用。在有碱金属盐和碱土金属盐、特别是锂盐存在的羰化反应体系中,碘甲烷对[Rh(CO)2I2]-的氧化加成亦是乙酸甲酯羰化反应过程的速度控制步骤。
尽管液相法络合催化羰化合成乙酸和乙酐工艺已在工业上获得了成功的应用,但对气相法络合催化羰化技术路线的研究仍然受到普遍的重视。与液相法相比,气相法是让呈气体状态的反应物在固载催化剂催化作用下实现羰化反应。这种气-固相反应在工艺上简化了反应流程,减轻了反应介质对设备的腐蚀程度,解决了液相法中产物与催化剂不易分离等诸多难题,且具有在更低压力下操作和生产效率高等优点。气相法络合催化羰化工艺采用的是负载型催化剂。作为负载型催化剂的主体金属,也是Ⅷ族过渡金属。就催化活性、选择性及反应的温和条件而论,仍然以金属铑为最佳选择。助催化剂为卤素或卤化物,亦如前述,以碘化物为最好。而作为负载型催化剂的载体,文献中报导有二氧化硅和金属(包括稀土)氧化物、沸石无机盐、碳质材料和活性炭,以及高聚物树脂等,其中以活性炭载体的效果为最好。J.F.Roth等人[39]将铑化合物负载在多孔活性炭上进行气相甲醇羰化实验,反应温度为208℃,压力1.6
MPa,催化反应速率270 mol AcOH/mol Rh.h,是氧化物载体的五倍以上。中科院成都有机化学所于20世纪70年代后半期和80年代初,曾就此进一步深入研究[40],将催化速率又提高到423
mol AcOH/mol Rh.h,并通过了1000小时以上的催化剂寿命考核实验。研究认为,在羰化催化剂中以活性炭作载体比沸石分子筛和金属氧化物的催化活性和选择性高,是由于活性炭的高比表面积及活性组分与载体之间的特殊相互作用,有利于表面催化反应中卤代甲烷对活性金属物种氧化加成的电子转移,从而促进羰化反应的进行[41]。在以活性炭为载体的含铑催化剂表面形成的催化活性物种很可能与均相铑催化剂催化羰化反应体系中的一样,即二羰基二碘铑(I)阴离子[Rh(CO)2I2]-或其它形式的一价铑Rh(I)活性物种类似物[40]。
采用Ⅷ族双金属或掺杂其它过渡金属的催化剂进行乙酸和乙酐的羰基合成,也一直有研究工作报道。壳牌国际研究所以乙酸钯-乙酸镍/碘化物和噻吩等在175℃和4
MPa压力下催化甲醇羰化制乙酸,催化速率达459 mol AcOH/mol Pd.h[42]。上述催化剂体系中大量加入乙酸镍(Ni/Pd比为8.5),羰化活性增加极为有限,若仅用镍盐,则催化速率仅及钯盐的2%左右。同篇报告的实验结果还指出,采用铂化合物Cl2Pt(PhCN)2取代乙酸钯,催化速率降到原来的0.5%以下。法国Rh?ne-Poulenc公司对Co-Ru[43]和Co-Fe[44]等双金属催化剂催化乙酸甲酯羰化反应进行了研究,反应温度210℃,压力25
MPa,发现实际效果不如Co-Cr催化剂[45]。中村和乾智将镍(8 wt%)和稀土氧化物(氧化镧5
wt%)负载在多孔硅胶上,并添加极少量铂族金属(0.1 wt%)进行甲醇与CO的气相反应,发现添加的金属对甲醇羰化产生乙酸甲酯有明显的协同催化作用[8,46],其活性顺序推断应当是:Rh>Ru>Pt>Pd>Ir>Ni。
络合催化羰化工艺用于乙酸和乙酐的羰基合成,近10多年来,在液相法和气相法两方面均取得了较大的新发展。
3 液相羰基合成及其催化技术的新发展
Hoechst-Celanese公司的Smith发现[47],在低含水量的反应体系中添加碘盐可实现如同高水浓度时一样的羰化反应速率,其原因在于碘盐可使反应体系中产生大量的乙酸根和碘负离子,并与[Rh(CO)2I2]-形成亲核能力更强、活性更高的五配位中间体[Rh(CO)
2I2(L)]-2 (L = I- 或AcO-),从而有利于提高氧化加成反应的速率。Hoechst-Celanese对Monsanto均相溶液催化剂体系的诸多改进,形成了一套所谓“酸优化”工艺,使反应体系中含水量可以大大降低,而催化反应速率却有所增加[48],达到2.6×103
mol AcOH/mol Rh.h,催化剂沉淀现象亦明显有所改善。
20世纪90年代,我们在高分子铑催化剂研究工作的基础上,发明了新一代用于均相或多相羰化反应的多齿季盐杂键合型铑离子配合物催化剂[49,50]。我们将一类分子骨架中既有季盐离子(L+)、又有自由碱基单元(Lo)的多齿有机或高分子化合物(L+~Lo)用作为配体分子,与铑的前体化合物、例如Rh2(CO)4Cl2或RhCl3.3H2O等反应形成杂键合型铑(I)离子配合物
[Rh(CO)2I2]- L+~Lo [51]。此处L+或写为L+(R),为季盐化的氮族元素基团,R为季盐化取代基。Lo为未季盐化的氮族元素碱性基团,但也可能是含具有孤对电子的氧族元素的基团。采用这类催化剂进行液相或液-固相羰化反应,就液相中的反应而论,其动力学速度方程与均相溶液的相同。由于在这类催化剂中,季盐离子L+(R)和自由碱基Lo两种配体单元在分子中同时并存且相互连接,分别可能以离子对和配位络合形式与[Rh(CO)
2I2]-相键合, 具有如下(A)和(A')互变的结构特征:
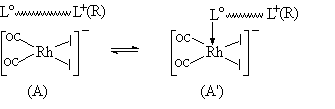
甲醇羰化实验表明,由于催化剂的配体分子中带有孤对电子的自由碱基(Lo),对于稳定对称的正方平面构型的二羰基二碘铑(I)阴离子中处于配位不饱和状态的铑活性中心、具有给电子配位络合作用,从而形成很不稳定、但活性更高的五配位中间体[Rh(CO)2I2(Lo)]-,其结果是诱发了卤代甲烷、特别是碘甲烷对铑催化活性物种的氧化加成,促进了羰化反应的进行,使催化活性显著地增强。以其中一种多齿季盐杂键合型铑催化剂为例,在120-185℃温度区间内进行甲醇羰化反应,由实验测定的反应活化能Ea和速度常数的指前因子k0分别为Ea=58.4
kJ. mol-1和k0=1.1×107 L.mol-1. sec-1,确定的反应活化热焓△H≠=54.9
kJ. mol-1,活化熵△S≠= -121.2 J.mol-1. K-1;而采用这种催化剂进行乙酸甲酯羰化反应,实验测定的反应活化参数分别为Ea=55.8
kJ. mol-1和k0=1.0×106
L.mol-1. sec-1;△H≠=52.3 kJ. mol-1,△S≠=
-141.1 J.mol-1. K-1,其催化活性为n-Bu4N+ -[Rh(CO)2I2]催化剂的2~6倍。采用这种类型的催化剂、特别是以高聚物作为配体时,时空产率可以更大幅度地得到提高。这是因为在这种催化剂体系中,铑活性物种能以比溶液中高出几十倍的浓度均匀地结合在高聚物分子链上。而在柔性高聚物基质中,催化反应的进行有可能做到与在均相溶液中的很接近,特别是当催化活性物种与高分子链段以恰当的多点位方式和杂键合类型结合时,则可以达到象生物酶那样充分发挥对反应物的作用。由于新型催化剂的配体分子设计集季盐配体单元和自由碱基配体单元于一身,这使配体分子及其形成的催化剂体系在多组分的不断变化着的羰化反应介质中的化学相容性能得到显著改善,同时也提高了反应连续相内部及相间的传质速度,其结果不但保持了现有的均相铑催化剂的高选择性,而且使羰化催化活性和稳定性、包括热稳定性得到了进一步的提高,从总体上改善了这类催化剂的综合使用性能。采用多齿季盐杂键合型铑催化剂由甲醇羰化合成乙酸,催化反应速率可以达到6.6×103
mol AcOH/mol Rh.h,羰化产物选择性保持在99%以上;而这类铑催化剂用于乙酸甲酯或二甲醚羰化合成乙酐,在反应温度170-180℃范围内,催化反应速率可达到2.8×103
mol AcOAc/mol Rh.h,羰化产物选择性接近100%。
20世纪90年代,BP公司实验发现[52,53],一种由VIII族铱(Ir)为主体金属构成的双金属元素催化剂对乙酸羰基合成有较好的催化效果。特别是由铱和钌(Ru)组成的VIII族双金属催化剂对乙酸羰基合成的催化效果更好,被称做Cativa催化剂[52,54]。在这类催化剂的羰化反应体系中,非铱金属羰基化合物被认为只是起催化促进剂的作用,催化活性物种与铑催化剂的类似,是[Ir(CO)2I2]-,只是碘甲烷对[Ir(CO)2I2]-的氧化加成速度是对[Rh(CO)2I2]-氧化加成速度的150倍[54]。研究指出,在采用铱催化剂的羰化反应体系中,羰化反应的速度决定步骤,已不再是碘甲烷对催化活性物种的氧化加成,而是其下一步反应,即从氧化加成产物中间体[Ir(CO)2I3Me]-中脱去一个碘负离子(I-),同时有一个外来羰基(CO)配位取代上去。如果以乙酸甲酯为甲氧基的主体原料来源,在合并考虑有机反应平衡后,则总体羰化反应速度
r = [Ir].[CO].[AcOMe]。Celanese公司报道[55],在使用均相铱催化剂进行乙酸羰基合成的羰化反应体系中,加入少量含吡啶环的交联聚合物,催化活性有明显改善,羰化反应速度一般能提高50%左右。数据表明,采用铱催化剂进行乙酸羰基合成所能达到的最好的催化反应速率为2.0×103
mol AcOH/mol Ir.h[54],与酸优化工艺使用铑催化剂的接近。采用铱催化剂进行乙酸羰基合成,其优点在于:⑴催化剂所用的贵金属为铱和钌,价格相对而言要比金属铑便宜一点;⑵作为助催化剂的碘甲烷的使用量,不像铑催化剂羰化反应体系中使用的那么多。但采用铱催化剂进行羰基合成,也有如下一些不足之处:⑴催化剂体系的组成比较复杂,各组成成分之间的相互依存性很强,故各组成成分之间的配比更要严格控制;⑵液体反应组分接触到的设备和管线中的金属腐蚀物、碱金属和碱土金属离子以及易于季盐化的含氮、磷原子基团的配体都有可能使铱催化剂的催化活性迅速降低,甚至失活。原因是它们会产生碘负离子(I-),故把它们统称为离子污染物。采用铱催化剂进行羰基合成,很重要的一项操作规程,就是要严格控制这些离子污染物在反应体系中的存在量。大家知道,在铱催化剂的催化循环过程中,又必须要有碘负离子的参与,只是其量不能多。有文献报道[53],碘负离子浓度应保持在500
ppm以下,最好是在250 ppm以下。而在铑催化剂的羰化反应体系中,基本上不存在所谓离子污染物影响之类的问题。
4 气相法羰基合成及其催化技术的新发展
尽管活性炭负载催化剂与以氧化物及沸石分子筛等为载体制备的催化剂相比,其催化性能要优越得多,但是活性炭作为载体也存在机械强度低、热稳定性不好和热传导性能差等一些严重的弱点,致使其在使用过程中容易失活,使用寿命短,很难满足气相法羰化合成乙酸在工业上连续生产的要求。造成催化剂失活的具体原因,分析有如下几个主要方面:(1)
活性炭的机械强度较低,耐热冲击性能较差,致使做成的催化剂在使用过程中,其构架容易发生破裂和坍塌;(2)
作为催化剂的活性组分金属铑,在载体表面迁移聚积造成有效活性表面积减小,导致活性降低;(3)
活性炭较疏松的体相结构,使其热导性能较差,由反应放热引起的局部过热现象使反应物容易在催化剂表面积焦,造成催化剂的比表面积及孔容大幅度地下降,以及积焦或积碳覆盖活性组分表面,更加速使催化剂失活。
20世纪90年代,我们尝试采用由不同高聚物树脂烧制成的碳多孔材料作载体,特别是开发了一类新型的以纤维状或多维结构的无机物填充增强的多孔碳复合材料(TFC)负载金属铑和稀土元素的催化剂(Rh/TFC)[11],用于气相法羰基合成,发现其使用效果比较理想。所用的载体是以无机物纤维、晶须或高维晶体粉末填充到酚醛树脂等高分子树脂中,制成高分子复合材料,经高温分解和炭化,再进行表面氧化和烧蚀扩孔等处理,制成多孔碳复合材料。以此作载体浸渍或沉积铑盐和稀土金属盐,再经还原和烧结处理,最终制得铑的负载型催化剂。由于新型催化剂在其载体中填充有无机物纤维、晶须或高维晶体粉末,其热稳定性、热导性能和机械强度等均得到显著改善;另外在载体制备过程中通过加入不同添加剂改进多孔载体的结构、特别是孔径结构分布,提高了最后制成的催化剂的传质和催化反应性能,以及综合使用性能。新型催化剂(Rh/TFC)的使用稳定性与活性炭负载型催化剂(Rh/A.C.)的相比有了很大程度的提高,其中最重要的是采用碳复合材料作为载体制成的催化剂在羰化反应过程中,积焦或积碳现象明显地趋于缓和,从而导致的比表面积下降和催化剂增重程度大大减轻。新型催化剂使用稳定性好的另一方面重要原因是,在羰化反应过程中催化剂的活性金属铑在载体表面的迁移聚积现象受到较大程度的抑制,这从催化剂使用前后的电镜照片和测定的反应产物中的铑含量得到了证实。在1000小时催化剂寿命考核实验过程中,连续抽样分析所测定的反应产物中的铑含量均小于10
ppb,而采用铑/活性炭催化剂进行气相甲醇羰化反应,检测到的反应产物中的铑流失量为70
ppb[40]。新型的以纤维状或多维结构无机物填充增强的多孔碳复合材料(TFC)负载金属铑和稀土元素的催化剂(Rh/TFC)用于气-固相络合催化羰化反应、由甲醇合成乙酸,在反应温度170-250℃、压力0.8~1.5
MPa范围内,在1000小时的连续催化反应过程中,甲醇转化率保持在60%以上,羰化产物的选择性高于99%,催化反应速率大于760
mol AcOH/mol Rh.h,时空产率在17 mol AcOH/L.h以上。
5 结束语
对于乙酸和乙酐羰基合成而言,尽管Koch碳正离子羰化反应被最先使用,但其实际效果较差。仅仅是近几年,在采用气相法Koch羰化反应合成乙酸领域有了一些显著的进步。而Reppe络合催化羰化反应从最初被发现时起,很快就被应用于乙酸和乙酐的工业生产,并且研究进展不断:特别是Monsanto的低压羰基合成铑催化剂的发现;近10多年来,在液相法羰基合成方面相继又有酸优化工艺、多齿季盐杂键合型铑催化剂、以及铱催化剂等推出;在气相法羰基合成方面,新型的铑/碳复合材料催化剂(Rh/TFC)体系开发成功,亦当属这一领域的一项重要进展。
REFERENCES
[1]蒋大智,李小宝,王恩来, 羰基合成化学(殷元骐主编),北京,化工出版社,1995,143-177。
[2]夏求真, 化工百科全书第2集 (陈冠荣主编), 北京,化工出版社,1991,717。
[3]J.Falbe, New Synthesis with Carbon Monoxide, Spring-Verlag, New york,1980.
[4]K.Fujimoto,I.Shikada,K.Omata and H-O.Tominaga, Chem.Lett., 1984, 2047-2050.
[5]B.Eills,J.Howrd,R.Joyner,et al.,11th ICC,101B,B-26,1996,771-779.
[6]Y.Souma, H.Sano, H.Miwa et al., 有机合成化学,1990,48(2),92-101.
[7]S.H.Vanderpool, Jiang-Jen Lin, R.G.Duranleau (Texaco Inc.),US 4 629 809 (1987).
[8]中村征四郎,乾智行, JP 56-104 838;JP 56-104 839 (1981).
[9]A.Calafat,J.Laine, Catalysis Letters, 1994, 28(1): 69-77.
[10]Feng Peng, J.natural Gas Chem., 2003, 12(1), 31-36.
[11]蒋大智,李小宝等,中国专利: CN 95 103 039(1995).
[12]Jiang-Jen Lin, R.G.Duranleau (Texaco Inc.), US 4 519 956 (1985).
[13]加藤 顺等编著,金革等译,《碳一化学工业生产技术》(1980), 北京,化工出版社,1988。
[14]W.H?lderich,G.Fouquet,F.C?sar(BASF),DE 3 606 169(1987).
[15]W.Reppe,H.F.Worms(BASF),US 2 729 651,US 2 730 546,US 2 789 137(1956).
[16]W.Reppe,H.Bille(BASF), Ger Pat. 1 006 847(1959).
[17]F.E.Paulik,J.F.Roth,Chem.Commun., 1968,24,1578.
[18]J.F.Roth,J.H.Craddock,A.Hershman,F.E.Paulik,Chem.Tech., 1971,10,600.
[19]F.E.Paulik,A.Hershman,J.F.Roth(Monsanto), US 3 769 329(1973).
[20]G.W.Adamson,J.J.Daly,D.Forster,J.Organomet.Chem. 1974,71,C17.
[21]D.Forster, J.Am.Chem.Soc. 1976,98,846.
[22]J.Hjortkjaer,V.W.Jensen,Ind.Eng.Chem.,Prod.Dev. 1976,15⑴,46.
[23] F.E.Paulik,T.C.Singleton,W.H.Urry(Monsanto), EP 055 618 (1982).
[24]王曼芳,厦门大学学报(自然科学版), 1986,25⑷,488-491.
[25]T.Kagama (Ajinomoto), JP 75-30 820(1973).
[26]T.Onouchi (Showa Denko), JP 75-47 922; JP 75-47 921(1973).
[27]N.Rizkalla and C.N.Winnick,Ger Pat.2 610 035(1976).
[28]C.Hewlett (Halcon),FR 2 242 362(1973).
[29]H.Erpenbach (Hoechst), FR 2 289 480(1974).
[30]H.Kuchertz, H.Erpenbach (Hoechst),Ger Pat. 2 450 965(1976).
[31]K.Gehrmann, H.K.Kubbeler (Hoechst),Ger Pat. 2 836 084(1980).
[32]J.E.Ehrler,B.Juran, Chemical Week,June 1(1983).
[33]松比良伸也,有机合成化学, 1984,42(2),115.
[34]Chee-Gen Wan(Halcon),US 4 234 719(1980).
[35]L.T.Hassell,P.S.Whitmore,T.G.Charles,Y.D.Alexander,JP 57-501 782(1982).
[36]V.A.Agreda,ChemTechnol., 1988,18,250.
[37]British Petroleum, EP 87 870(1983).
[38]G.Luft,M.Schrod, J.Mol.Catal. 1983,20,175-184.
[39]K.K.Robinson,A.Hershman,J.H.Craddock,J.F.Roth,J.Catal.,1972,27,389.
[40]中科院成都有机化学所,石油化工,a) 1980,9⑽,573; b) 1981,10⑸,301.
[41]K.Omata,K.Fujimoto,et al.,Ind.Eng.Chem.Prod.Res.Dev.,1985,24,234-239.
[42]P.W.N.M.Van Leeuwen,C.F.Roobeek, NL 2 644(1983).
[43]J.Gauthier-Lafaye,R.Perron(Rh?ne-Poulenc),EP 067 777(1982).
[44]C.Doussain,J.Gauthier-Lafaye,R.Perron(Rh?ne-Poulenc),EP 070 787(1983).
[45]J.Galluci,J.Gauthier-Lafaye,R.Perron(Rh?ne-Poulenc),EP 070 788(1983).
[46]中村征四郎,乾智行,JP 59-053 440(1984).
[47]B.L.Smith,J.Mol.Catal.,1987,39,115-136.
[48]B.L.Smith,G.P.Torrence,A.Aguilo,J.S.Alder, US 5 026 908(1991).
[49]蒋大智,李小宝,王恩来等,中国专利: CN 95 108 227(1995).
[50]蒋大智,王恩来,李小宝等,中国专利: CN 95 104 298(1995).
[51]Dazhi Jiang,Xiaobao Li,Enlei,Wang,Macromol.Symp., 1996,105,161-166.
[52]S.J.Glenn,D.E.Jan,W.R.John, EP 643 034(1995); EP 849 248(1998).
[53]G.M.Francis,R.Georgios,B.M.James,G.C.Sherman, EP 752 406/749 948(1996).
[54]J.H.Jones,Platinum Metals Rev., 2000,4(3),94-1105.
[55]Hung-Cheun Cheung,E.C.Sibrel,G.P.Torrence, US 6 458 995(2002).