http://www.chemistrymag.org/cji/2005/075034pc.htm |
May 2, 2005 Vol.7 No.5 P.34 Copyright |
Peng Chenfeng, Zhang Yuanming, Tang Yu, Li Hong, Liu Yingliang
( Department of Chemistry,Ji'nan University,Guangzhou
510632)
Keyword Titania; Phosphotungstic acid; Photocatalysis; Solvothermal synthesis 溶剂热法制备磷钨酸/二氧化钛光催化剂及性能研究
彭晨凤,张渊明,唐渝,李红,刘应亮
(暨南大学化学系,广州,510632)
2005年3月16日收稿; 国家自然科学基金资助项目(编号:20171018)
摘要 采用溶剂热法在120℃下制备了复合光催化剂H3PW12O40/TiO2(记作PW12/TiO2)。利用FT-IR、XRD、TEM对其进行了表征。以光催化降解甲基橙为探针反应,考察磷钨酸的掺杂量、催化剂用量、甲基橙溶液初始浓度等因素对光催化剂活性的影响。结果表明:PW12/TiO2颗粒尺寸为30-50
nm;XRD证明PW12/TiO2光催化剂中的TiO2为锐钛矿相,H3PW12O40阴离子的Keggin结构仍然保持;PW12/TiO2具有较高的光催化活性,是Deguassa
P-25 TiO2的7.0倍,是直接负载了磷钨酸的P-25光催化剂的4.1倍。不同光催化剂的活性依次为:PW12/TiO2>
PW12/P-25> P-25>TiO2。在1.2 g/L的PW12/TiO2存在下光照10
min,250 ml 20 mg/L甲基橙溶液完全降解。
关键词 二氧化钛 磷钨酸 光催化 溶剂热法
光催化氧化技术能彻底将有机污染物分解为CO2、H2O及其它无机小分子,在处理难生物降解的有机污染物方面具有广阔的应用前景[1]。其中,TiO2以带隙能适中、高光催化活性、光稳定性和化学稳定性等优点而研究最为广泛。近年来,TiO2光催化剂通过改性如贵金属沉积、金属离子掺杂、二元或多元半导体复合等方法[2
],提高TiO2的光催化活性成为研究热点。杂多酸具有类似半导体氧化物能带结构,其光催化性能得到广泛关注[3]。杂多酸对TiO2修饰的研究也处于探讨之中[4]。将杂多酸直接加入到TiO2光催化反应体系中虽然能提高TiO2光催化活性[5,6],但杂多酸在该体系中以均相存在,不利于催化剂的回收利用;如果将杂多酸直接负载在TiO2[4]上,在使用过程中杂多酸又容易脱落。李丹峰[7]等人采用sol-gel法制备杂多酸/TiO2复合催化剂,但催化剂需要高温热处理晶化,容易造成杂多酸分解。胡长文[8]等采用纳米尺度多孔锐钛矿TiO2与磷钨酸在200
℃下形成了H3PW12O40/TiO2复合物,在可见光下显示出较好的光催化降解活性。我们采用溶剂热法在更低的温度(120℃)下制备磷钨酸/TiO2复合催化剂,其稳定性高,且TiO2晶化程度好,具有较高的光催化活性,比直接将磷钨酸负载在TiO2上的光催化活性高。
1 实验部分
1.1 试剂及仪器
P-25 TiO2 粉末(德国Degussa
公司商品),甲基橙(AR,湖南金涛化学试剂研究所),十二磷钨酸H3PW12O40(AR,上海润捷化学试剂有限公司),钛酸丁酯为化学纯,其它试剂为分析纯。
Spectrumlab 22pc 可见分光光度计(Lengquang Tech.)测定溶液中甲基橙浓度;PHS-3C精密pH计测量溶液pH值;红外光谱采用Bruker
EQUINOX 55 傅立叶变换红外光谱仪,KBr压片; X-ray 衍射图谱由MSAL XD-2
型 (Cu Ka,40 kV,20mA)X-ray 粉末衍射仪得到,2θ 角 从20o到70o。透射电子显微镜(TEM)
PHILIPS TECANAI 10
1.2 HPA/TiO2复合催化剂的制备
8 ml钛酸丁酯搅拌下加入到60 ml乙醇中,溶解均匀后加入一定量的HNO3溶液(与水的体积比为1:5),反应一定时间后,滴加5 ml磷钨酸的乙醇溶液(制备纯TiO2则省略该步),搅拌几小时后,装入加有聚四氟乙烯内衬的筒式高压釜中在120℃下反应24 h。产物分别用丙酮、水洗涤两次,在80℃下真空干燥12 h,然后研磨得TiO2或PW12/TiO2粉体。以P-25代替钛酸丁酯作为钛源,制备了PW12/P-25复合物。
1.3光催化实验
光催化实验是在自制的可控温反应器中进行,光源为125 W汞灯(上海亚明GGZ-125),与液面垂直距离为10 cm,连续搅拌并鼓入空气 (空气流速1.7 L/min)。甲基橙初始浓度为20 mg/L,体积为250 ml,加入一定量的催化剂,反应前在黑暗下搅拌0.5 h。每隔一段时间抽取一定量的溶液,经离心分离后取上层清液用分光光度计测量其吸光度。在本实验所用浓度范围甲基橙溶液吸光度与浓度成线性关系。
2 结果与讨论
2.1 催化剂的表征
图1为所制催化剂的红外图谱。从图中可以看出,在700-500 cm-1的吸收峰为TiO2的Ti-O振动峰。PW12/TiO2复合物在1100-750 cm-1保留有Keggin结构杂多阴离子的四个特征吸收带,杂多阴离子基本骨架结构未遭破坏。金属边氧键M-Oe-M和金属角氧键M-Oc-M的伸缩振动吸收峰分别由807 cm-1和889 cm-1红移到817 cm-1和895 cm-1。同时,金属端氧键M-Ot和磷与内氧键P-Op的伸缩振动吸收峰分别由984cm-1和1080 cm-1蓝移到967 cm-1 和1071 cm-1。这可能是因为在复合材料的制备过程,随着钛酸丁酯在酸性条件下的水解,不断生成TiO2网络,体积庞大的PW12O403-进入TiO2网络中,其结果是导致杂多阴离子被牢固地束缚在TiO2网络[9],杂多阴离子与TiO2网络之间相互作用使得杂多阴离子的特征吸收带产生位移。
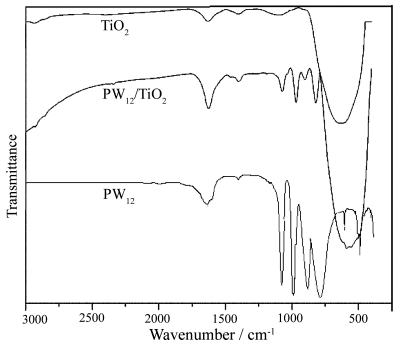
图1 各种催化剂的FT-IR 图谱
Fig.1 FT-IR spectra of TiO2, PW12/TiO2 and PW12
图2为样品的X射线粉末衍射图谱。从图中可以明显观察样品TiO2、PW12/TiO2在2q分别为25.2,38.1,48.3,54.5,62.5°处有衍射峰,它们是TiO2锐钛矿相特征衍射峰。表明样品中的TiO2是以晶体形式存在的。通常锐钛矿相TiO2光催化剂比其它晶相如金红石,表现出更好的光催化活性。常用的溶胶凝胶法合成锐钛矿相TiO2需要在500℃左右进行焙烧,本工作采用溶剂热法,在120℃下就能够实现,这符合溶剂热条件下晶体生长的一般规律。而这个结果对于将杂多化合物与TiO2组成复合材料,同时保持杂多阴离子原来的结构是非常重要的。因为杂多阴离子的热稳定性不高,经过400℃左右的热处理,杂多化合物随即分解,这也是杂多化合物与其它物质难以形成复合材料的主要原因之一。图2中没有出现PW12的特征衍射峰,可能是磷钨酸以弥散状态存在,并被束缚在TiO2网络中
[10]。
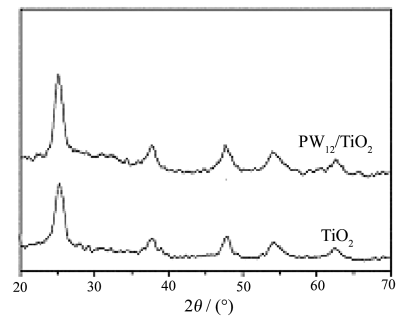
图2 TiO2 和 PW12/TiO2的XRD 图谱
Fig.2 XRD patterns of TiO2 and
PW12/TiO2

图3 PW12/TiO2的TEM图像
Fig. 3 TEM image of PW12/TiO2
2.2 光催化性能研究
2.2.1不同催化剂的光催化活性比较
固定甲基橙溶液的初始浓度为20 mg/L,催化剂的投入量为1.2
g/L,选用不同的催化剂,于相同条件下比较甲基橙的脱色速率(图4)。从图中可以看出,使用PW12/TiO2复合催化剂,在10
min内,甲基橙就基本降解完全,而PW12/ P25、纯P-25则需要将近35
min。在本研究的反应条件,按照简单的一级反应动力学关系处理数据,得出在相同的实验条件下,PW12/TiO2
(磷钨酸,21.01% ) 光催化反应速率(k)是P-25 TiO2的7.0倍,是直接负载的磷钨酸/P-25光催化剂的4.1倍,PW12/TiO2的催化效果明显好于相同方法制备而不加入磷钨酸的纯TiO2,以及用P-25代替钛酸丁酯制备的PW12/P25,还有P-25,显示所制备的复合催化剂中磷钨酸与TiO2之间存在协同效应。
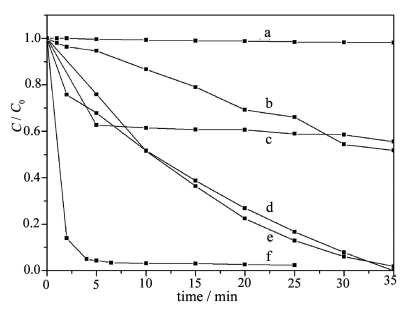
图4 不同催化剂的光催化降解甲基橙活性
Fig.4 Residual chroma of methyl
orange solution degradated by different catalysts
(a) catalyst free, (b) H3PW12O40,
(c) TiO2, (d) PW12/P2-5, (e) P-25, (f) PW12/TiO2
(0.3g catalyst, w (PW12)%=21.01%, 250 ml 20 mg/L methyl orange solution)
2.2.2 磷钨酸对TiO2光催化效率的影响
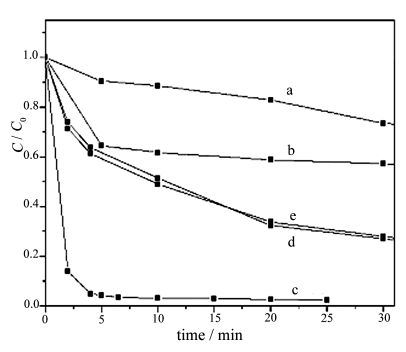
图5 不同磷钨酸掺杂量的PW12/TiO2光催化速率比较
Fig.5 Comparison of
photocatalytic rate on PW12/TiO2 with different doping amounts of H3PW12O40 w(PW12)%:(a) 0.00, (b) 9.62, (c) 21.01, (d) 29.85, (e)34.72
图5是磷钨酸掺杂量对催化剂光催化效率的影响。PW12/TiO2复合催化剂投入量为1.2g/L。从图中可以看出,磷钨酸的掺杂量的质量百分含量从0%增加到21.01%时,光催化反应速率显著增加,光催化反应速率常数(k)从0.0121
min-1提高到0.7509 min-1。磷钨酸的加入显著提高了TiO2光催化活性,这是因为磷钨酸具有与半导体金属氧化物相似之处,它的最低空轨道(LUMO)与最高占有轨道(HOMO)能级差为2.9-3.0
eV[11]。它本身能在光的激发下,产生光生空穴-电子对,催化氧化有机污染物。另一方面,PW12O403-是一个很好的电子接受体,每个分子能接受多个电子[12-14](式1)。
![]()
在PW12/TiO2复合催化剂中,磷钨酸被牢固地束缚在TiO2网络,且有合适的能隙结构,能迅速使TiO2所产生的光生电子转移,从而减少空穴与电子直接复合,提高光降解甲基橙效率。在O2存在下[15],PW12O404-可以被氧化为PW12O403-。磷钨酸在这个吸收/释放电子过程中,可以看作电子转移介质,将光生电子从TiO2传递到O2
( 式2 )。
![]()
由于磷钨酸与TiO2原位形成复合物,两者的结合较一般的机械混合或负载更强,间接地将电子转移到溶解氧的速率也大大提高了。同时,PW12O403-作用类似如过渡金属离子修饰TiO2,在TiO2晶格里形成俘获光生电子陷阱,从而提高了光催化反应速率。另一方面,PW12/
P25之间的结合是在酸性的条件下实现的,TiO2表面电荷呈正电荷,带负电荷的PW12O403-离子通过静电力的作用吸附在TiO2表面。但如果两者之间仅仅是表面的物理吸附,如磷钨酸与P-25,它们的协同催化效果并不明显,其光催化降解能力比PW12/TiO2要差。这也进一步说明PW12/TiO2复合物中,磷钨酸与TiO2之间不仅仅是简单的物理吸附。
当掺杂量大于21.01%时,继续增磷钨酸掺杂量,光催化反应速率反而下降,在磷钨酸的掺杂量增加到44.91%时,k下降为0.0791min-1。这是因为磷钨酸(HPW)掺杂量过多时,磷钨酸在TiO2表面及晶格里面也相对多一些。在TiO2表面HPW量的增加而导致HPW更多地占据了TiO2表面的活性中心,作为光催化剂的TiO2与模拟污染物甲基橙的接触面积减少,光催化反应速率也下降;另一方面,TiO2晶格里面作为电子陷阱的HPW也有可能因为过量而充当光生空穴-电子的复合中心,也会降低光催化反应活性。
2.2.3 催化剂用量对反应的影响
图6为不同催化剂加入量对甲基橙脱色速率的影响。实验结果表明,从0.4
g/L到2.0 g/L,光催化反应速率存在一个最佳值 [10]。在本实验条件下,PW12/TiO2用量为1.2g/L时,光催化反应速率达到最大,k为0.7509
min-1,在此条件下光照10 min,250 ml 20 mg/L甲基橙溶液完全降解。当催化剂加入量少时,光源产生的光子不能被充分利用。这种情况会因为催化剂用量的增加而有所改善,催化剂量的增加对光子的吸收及染料分子的吸附也增加,因而光催化反应速率也有所提高。当催化剂用量过多时,光催化剂容易发生聚集而沉降,降低了光催化剂的总比表面积,同时光催化颗粒对入射光产生散射使它对光利用率下降,从而导致光催化效率下降。
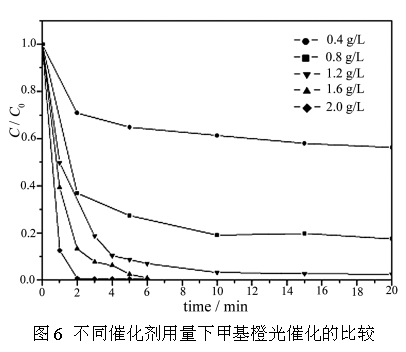
图6 不同催化剂用量下甲基橙光催化的比较
Fig.6 The comparison of
photocatalysis rate of methyl orange with different catalyst loadings w(PW12)%:21.01% , 250 ml 20 mg/L methyl orange solution
2.2.4 甲基橙起始浓度对反应的影响
图7为不同起始浓度下,甲基橙浓度的相对降低值c/c0与光照时间的变化关系。从图中可以看出,随着甲基橙浓度的增加,光催化反应速率降低。甲基橙的浓度从15
mg/L增加到30 mg/L时,光催化反应速率常数从0.8677 min-1降低到0.1159
min-1。这是因为随着甲基橙起始浓度的增加,紫外光透过反应溶液的能力下降,光催化剂对光的利用率也下降。另一方面光催化反应的动力学方程符合Langmuir-
Hinshelwood模式,其方程为:
![]()
当甲基橙浓度增大时,K[c]值大,光催化反应速率降低;当甲基橙浓度较小时,K[c]项可以忽略,此时反应可近似看作是一级反应。因此,在应用光催化降解有机污染物技术于时,选择合适的污染物浓度范围,对于光催化反应装置的设计及光催化处理废水效率是至关重要的。
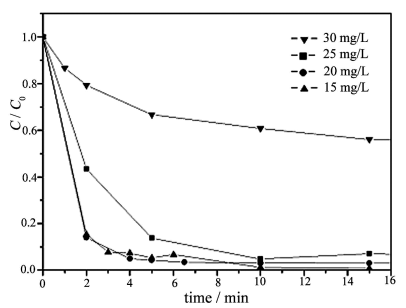
图7 甲基橙起始浓度对光降解速率的影响
Fig.7 The effect of intial concention
of methyl orange on photocatalysis rate
(0.3 g catalyst,w(PW12)%=21.01%,250 ml methyl orange)
3 结论
采用溶剂热法在120℃下制备了复合光催化剂H3PW12O40/TiO2并利用FT-IR、XRD、TEM对其进行了表征。PW12/TiO2颗粒为纳米尺度,TiO2为锐钛矿相,H3PW12O40阴离子的Keggin结构仍然保持,因此本文为低温下制备含杂多酸的复合光催化剂提供了一个新途径。光催化降解甲基橙反应结果显示:杂多酸与TiO2之间存在着明显的协同效应,PW12/TiO2的光催化活性高,是P-25
TiO2的7.0倍,是直接负载了磷钨酸的P-25光催化剂的4.1倍。不同光催化剂的活性依次为:PW12/TiO2>
PW12/P-25> P-25>TiO2。
REFERENCES
[1] Shi Y P, Yang Z H, Feng X, et al. Chinese Journal
of Catalysis (Cuihua Xuebao), 2003, 24 (9): 663.
[2] Wang C Y, Liu C Y, Shen T. Chemical Journal of Chinese University
(Gaodengxuexiao Huaxuexuebao), 1998, 19: 2013.
[3] Hiskia a, Ecke M,Troupis A, et al. Environ. Sci. Technol., 2001, 35: 2358.
[4] Anandan S, Yoon M. J. Photochem. and Photobio. C, 2003, 4 (1): 5.
[5] Yoon M, Chang J A, Kim Y, et al. J. Phys. Chem. B, 2001, 105: 2539.
[6] Lee H, Choi W. Environ. Sci. Technol., 2002, 36: 3872.
[7] Li Danfeng. Ph.D. thesis, Northeast Normal University, 2003: 8.
[8] Yang Y, Wu Q Y, Hu C W, et al. J. Mol. Catal. A: Chem., 2005, 225 (2): 203.
[9] Li D F, Guo Y H, Hu C W, et al. Appl. Catal. A: General, 2002, 235: 11.
[10] Yan P F, Zhou D R, Wang J Q, et al. Chemical Journal of Chinese University
(Gaodengxuexiao Huaxuexuebao), 2002, 23 (12): 2317.
[11] Friesen D A, Headley J V, Langford C H. Environ. Sci. Technol., 1999, 33: 3193.
[12] Na K, Okuhara T, Misono M. J. Catal., 1997, 170: 96.
[13] Hill C L, Bouchard D A. J. Am. Chem. Soc., 1985, 107: 5148.
[14] Akid R, Darwent J R. J. Chem. Soc. Dalton Trans., 1985: 395.
[15] Androulaki E, Hiskia A, Dimotikali D, et al. Environ. Sci. Technol., 2000, 34 (10):
2024.